Abstract
Somatostatin receptors (SSTRs) are variably expressed by a variety of malignancies. Using radiolabeled somatostatin analogs (SSAs), the presence of SSTRs on tumor cells may be exploited for molecular imaging and for peptide receptor radionuclide therapy. 111In-DTPA-octreotide has long been the standard in SSTR scintigraphy. A major leap forward was the introduction of gallium-68 labeled SSAs for positron emission tomography (PET) offering improved sensitivity. Tracers currently in clinical use are 68Ga-DOTA-Tyr3-octreotide (68Ga-DOTATOC), 68Ga-DOTA-Tyr3-octreotate (68Ga-DOTATATE) and 68Ga-DOTA-1-NaI3-octreotide (68Ga-DOTANOC), collectively referred to as 68Ga-DOTA-peptides. 68Ga-DOTA-peptide PET has superseded 111In-DTPA-octreotide scintigraphy as the modality of choice for SSTR imaging. However, implementation of 68Ga-DOTA-peptides in routine clinical practice is often limited by practical, economical and regulatory factors related to the use of the current generation of 68Ge/68Ga generators. Centralized production and distribution is challenging due to the low production yield and relatively short half-life of gallium-68. Furthermore, gallium-68 has a relatively long positron range, compromising spatial resolution on modern PET cameras. Therefore, possibilities of using other PET radionuclides are being explored. On the other hand, new developments in SSTR PET ligands are strongly driven by the need for improved lesion targeting, especially for tumors with low SSTR expression. This may be achieved by using peptide vectors having a higher affinity for the SSTR or a broader affinity profile for the different receptor subtypes or by using compounds recognizing more binding sites, such as SSTR antagonists. This review gives an overview of recent developments leading to the next generation of clinical PET tracers for SSTR imaging.
Introduction and background
Somatostatin receptors (SSTRs) are G-protein coupled membrane receptors that were first described in rat pituitary tumor cells by Schonbrunn and Tashjian in 1978 [1]. Five different human subtypes have been identified, named SSTR1 to 5 [2]. While the genes for SSTR1, 3, 4 and 5 are intronless, the SSTR2 gene produces two splice variants, SSTR2A and B, differing only in the length of their cytoplasmic tail [3,4]. SSTRs are expressed by a wide variety of normal human tissues, both in various regions of the brain and peripheral organs, such as the spleen, adrenals, pituitary gland, pancreas, liver, gastro-intestinal tract, kidneys and lungs, each exhibiting a characteristic expression pattern of the different SSTR subtypes [5-8]. SSTRs have also been identified in several human tumor types. Neuroendocrine tumors (NETs) represent one of the groups with the highest incidence of SSTR expression [9]. For instance in gastroenteropancreatic (GEP) NETs, SSTRs are present in 80 to 100% of cases, except for insulinomas, which have a lower incidence of 50 to 70% [10]. Other NETs expressing SSTRs include pituitary adenomas, pheochromocytomas, paragangliomas, lung carcinoids, small-cell lung cancers, Merkel cell carcinomas, medullary thyroid carcinomas and neuroblastomas [11]. A wide variability in receptor density and subtype expression has been observed across the different NET types, but also within individual tumor types [9,11]. In the majority of cases, SSTR2 is most abundant, even when other subtypes are present [8,12]. A large variety of other solid and hematological malignancies may also variably express SSTRs. These include meningiomas, gliomas, lymphomas, and breast, lung, renal cell, pancreato-biliary tract, liver cell, colorectal, ovarian and prostatic carcinomas [9,11,12].
The presence of SSTRs on tumor cells may open up an important window of opportunity for the clinical management of those tumors in terms of imaging and therapeutic options. Especially in NETs this opportunity has already been extensively exploited. SSTR overexpression is the foundation on which the use of somatostatin analogs (SSAs) such as octreotide in the pharmacological treatment of NETs is based [13]. According to the most recent European Neuroendocrine Tumor Society (ENETS) consensus guidelines, SSAs are indicated for symptom relief in case of functioning tumors that cause hormone production, as well as for tumor growth inhibition [14].
Furthermore, SSAs can be labeled with radionuclides. These radionuclides either have a heavy nucleus (Z > 83) or possess an imbalance in proton/neutron ratio, or are in a metastable energy state and will undergo radioactive decay. The excess of energy in the nucleus of the unstable element can result in emission of either particles (α, β+/-) and/or electromagnetic radiation (gamma ray photons (γ)) and as a secondary effect X-rays, conversion electrons and Auger electrons. The specific decay characteristics of the radionuclide attached to the vector molecule determine if the radiopharmaceutical can be used for diagnostic (molecular imaging) or therapeutic (targeted radionuclide therapy) purposes. As such, SSTR imaging now occupies a key position in the clinical management of NETs [15-17]. Moreover, radiolabeling of SSTR targeting agents with therapeutic radionuclides may allow for vectorized radionuclide therapy, also called peptide receptor radionuclide therapy (PRRT). Currently, PRRT by means of radiolabeled SSAs represents an established, evidence-based treatment modality in case of inoperable/metastatic well-differentiated NETs [18] and its role has been enforced by the excellent results obtained in the randomized, controlled NETTER-1 trial [19].
In this review, we will focus on molecular imaging of the SSTR and more specifically on the developments leading to the next generation of clinical PET tracers targeting the SSTR. Peptides can be radiolabeled with radiohalogens (e.g. iodine isotopes and fluorine-18) by standard carbon-halogen bond formation or with radiometals (e.g. indium-111 and gallium-68) using suitable bifunctional chelators that are covalently linked to the biologically active peptide [20]. Therefore, peptide-based radiopharmaceuticals typically consist of the vector moiety (the biologically active peptide) which is linked by means of a chelator, and possibly through an additional linker, to the radionuclide [21]. Figure 1 shows the chemical structure of all relevant chelators discussed in this review with their matching radionuclides, in combination with the different SSAs that have been applied in clinical practice or clinical studies. Crucial for an effective SSTR tracer is its potential to bind the relevant SSTR [22]. It is important to realize that even small changes in the amino acid sequence of the peptide or a different choice of chelator or radionuclide might result in a different affinity profile [22,23] (see also Tables 1 and and2).2). Recent advances in molecular imaging of the SSTR, relating to the choice of radionuclide and vectors molecules, will be discussed. Figure 2 gives an overview of the likely future directions in the field of clinical SSTR PET imaging.

Chemical structures of the relevant SSTR tracers that have been applied in clinical practice or clinical studies: radionuclides with their matching chelators or fluorine-18 labeled constructs plus somatostatin receptor agonists or antagonists. (BASS: pNO2-Phe-c(D-Cys-Tyr-D-Trp-Lys-Thr-Cys)D-TyrNH2; JR11: Cpa-c[D-Cys-Aph(Hor)D-Aph(Cbm)-Lys-Thr-Cys]-D-Tyr-NH2).

Overview of the likely future directions in the field of clinical SSTR PET imaging.
Table 1
In vitro affinity profile (50% inhibitory concentration (IC50) in nM ± standard error of the mean) for the human somatostatin receptor of several somatostatin analogs
| Somatostatin analog | SSTR1 | SSTR2 | SSTR3 | SSTR4 | SSTR5 | Data from |
|---|---|---|---|---|---|---|
| Octreotide | > 10,000 | 2.0 ± 0.7 | 187 ± 55 | > 1,000 | 22 ± 6 | [23] |
| In-DTPA-octreotide | > 10,000 | 22 ± 3.6 | 182 ± 13 | > 1,000 | 237 ± 52 | [23] |
| Ga-DOTATATE | > 10,000 | 0.2 ± 0.04 | > 1,000 | 300 ± 140 | 377 ± 18 | [23] |
| Ga-DOTATOC | > 10,000 | 2.5 ± 0.5 | 613 ± 140 | > 1,000 | 73 ± 12 | [23] |
| Ga-DOTANOC | > 10,000 | 1.9 ± 0.4 | 40 ± 5.8 | 260 ± 74 | 7.2 ± 1.6 | [35] |
| Gluc-Lys-FP-TOCA | > 10,000 | 2.8 ± 0.4 | > 1,000 | 437 ± 84 | 123 ± 8.8 | [125] |
| F-FET-βAG-TOCA | NA | 4.7 | NA | 8,600 | NA | [69] |
| DOTA-lanreotide | > 10,000 | 26 ± 3.4 | 771 ± 229 | > 10,000 | 73 ± 12 | [23] |
| Y-DOTA-lanreotide | > 10,000 | 23 ± 5 | 290 ± 105 | > 10,000 | 16 ± 3.4 | [23] |
| Ga-KE88 | 9.5 ± 4.3 | 4.1 ± 1.4 | 2.7 ± 1.0 | 4.9 ± 1.4 | 2.25 ± 0.5 | [102] |
| AM3 | 119 ± 6 | 2.3 ± 0.2 | 4.0 ± 0.03 | 97 ± 21 | 27 ± 1 | [103] |
| DOTA-LTT-SS28 | 9.8 ± 0.2 | 2.5 ± 0.3 | 2.2 ± 0.5 | 4.8 ± 1.1 | 2.8 ± 0.3 | [105] |
| In-DOTA-LTT-SS28 | 14 ± 1.2 | 1.8 ± 0.2 | 4.0 ± 0.2 | 5.4 ± 0.3 | 1.4 ± 0.2 | [105] |
| DOTA-BASS | > 1,000 | 1.5 ± 0.4 | > 1,000 | 287 ± 27 | > 1,000 | [110] |
| In-DOTA-BASS | > 1,000 | 9.4 ± 0.4 | > 1,000 | 380 ± 57 | > 1,000 | [110] |
| NODAGA-JR11 | > 1,000 | 4.1 ± 0.2 | > 1,000 | > 1,000 | > 1,000 | [114] |
| Ga-NODAGA-JR11 | > 1,000 | 1.2 ± 0.2 | > 1,000 | > 1,000 | > 1,000 | [114] |
| DOTA-JR11 | > 1,000 | 0.72 ± 0.12 | > 1,000 | > 1,000 | > 1,000 | [114] |
| Ga-DOTA-JR11 | > 1,000 | 29 ± 2.7 | > 1,000 | > 1,000 | > 1,000 | [114] |
| Lu-DOTA-JR11 | > 1,000 | 0.73 ± 0.15 | > 1,000 | > 1,000 | > 1,000 | [114] |
Structures are explained in Figure 1, except for KE88 (DOTA-D-Dab-Arg-Phe-Phe-D-Trp-Lys-Thr-Phe), AM3 (DOTA-Tyr-cyclo(DAB-Arg-cyclo(Cys-Phe-D-Trp-Lys-Thr-Cys))) and SS28 (somatostatin-28).
Table 2
IC50 values (in nM ± standard error of the mean (SEM)) from competitive binding assays in the rat pancreatic cancer cell line AR42J with high SSTR2 expression for several somatostatin analogs
From SPECT to PET. SSTR imaging: current status
SSTR imaging was first performed in humans in the late 1980s using 123I-Tyr3-octreotide [24]. However, due to several disadvantages such as a cumbersome radiolabeling procedure, high cost, limited availability of Na123I, and considerable amount of intestinal accumulation of activity complicating interpretation of images, iodine-123 was soon replaced by indium-111, bound to the peptide by means of the chelator diethylenetriaminepentaacetic acid (DTPA) [25,26]. 111In-DTPA-octreotide (or 111In-pentetreotide) has long been the standard in SSTR imaging [22,27]. Indium-111 is a γ-emitting radionuclide, thus imaging is performed by means of planar scintigraphy or single photon emission computed tomography (SPECT), with or without computed tomography (CT). However, there are some drawbacks to the use of indium-111, such as unfavorable nuclear physical characteristics resulting in suboptimal image quality and relatively high effective doses, limited availability and high costs [27,28]. Successful efforts have been made to label SSAs with technetium-99m instead [27-29]. One of these compounds is 99mTc-ethylenediamine-N,N’-diacetic acid/hydrazinonicotinamide-Tyr3-octreotide (99mTc-EDDA/HYNIC-TOC) (see Figure 1), which is registered in Poland (99mTc-Tektrotyd; Polatom) and used in several mainly Eastern European countries.
A major leap forward was the introduction of SSAs labeled with the positron-emitting radionuclide gallium-68 for positron emission tomography (PET) applications. This was made possible by the development of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), a macrocyclic chelator capable of forming stable complexes with a multitude of 2+ and 3+ charged radiometals [30,31] that can be coupled to SSAs (see Figure 1). PET offers several advantages over SPECT, such as a higher sensitivity and spatial resolution and the possibility for straightforward image quantification [22]. The first clinical publication in this field reported on the use of 68Ga-DOTA-TOC (68Ga-DOTATOC) in patients with meningiomas [32], closely followed by a publication on the application of 68Ga-DOTATOC in NET patients [33], both in 2001. Since then, 68Ga-labeled SSA PET has rapidly emerged as an established technique for SSTR imaging, especially in NETs where it represents the molecular imaging modality of choice [17,34]. Tracers currently in clinical use are 68Ga-DOTATOC, 68Ga-DOTA-Tyr3-octreotate (68Ga-DOTATATE) and 68Ga-DOTA-1-NaI3-octreotide (68Ga-DOTANOC), collectively referred to as 68Ga-DOTA-peptides (see Figure 1).
Table 1 shows the affinity profiles, determined by means of in vitro binding studies, of most of the SSTR PET ligands discussed in this review as compared to 111In-DTPA-octreotide and octreotide. All listed 68Ga-DOTA-peptides show higher affinity for SSTR2 than 111In-DTPA-octreotide [23,35]. Therefore, in tumors where SSTR2 is the most overexpressed subtype, such as NETs, this offers an additional benefit on top of the physical advantages associated with PET. As such, smaller lesions as well as lesions with low-to-moderate SSTR expression can be detected using 68Ga-DOTA-peptide PET imaging versus 111In-DTPA-octreotide SPECT [15]. Indeed, several studies comparing both techniques in a head-to-head manner consistently reported a superior performance of 68Ga-DOTA-peptide PET in NET patients [36-44] (see Table 3). An example is shown in Figure 3. An additional advantage is the fact that 68Ga-DOTATATE and 68Ga-DOTATOC are the theranostic twins of 177Lu-DOTATATE and 90Y-DOTATOC, currently the most frequently used radiopharmaceuticals for PRRT, and are as such ideally suited to identify eligible patients [45].

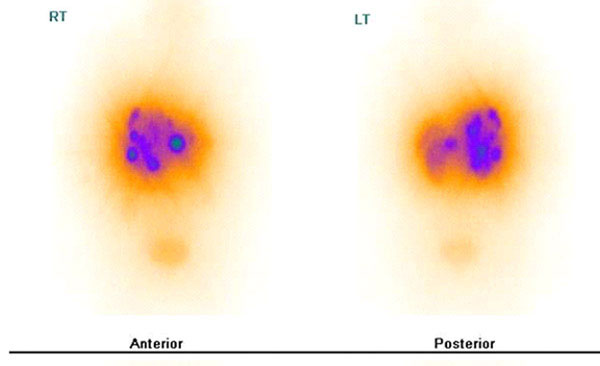
Head-to-head comparison of 111In-DTPA-octreotide scintigraphy (A: planar anterior, B: planar posterior, C: transversal SPECT image) and 68Ga-DOTATOC PET (D: maximal-intensity projection, E: transversal slice) of a patient with ileal NET and liver, lymph node and peritoneal metastases (patient data from: [41]). More lesions can be visualized on the 68Ga-DOTATOC PET images. Dashed lines in (A, B and D) denote the level of transversal slices in (C and E). Scale bar applies to PET images. (SUV = standardized uptake value).
Table 3
Comparison between the sensitivity of 111In-DTPA-octreotide planar and/or SPECT and 68Ga-DOTA-peptide PET as reported in several publications
| Study | n | Gallium-68 peptide | Analysis level | Sensitivity 111In-DTPA-octreotide | Sensitivity 68Ga-DOTA-peptide | Δ |
|---|---|---|---|---|---|---|
| Gabriel et al. [37] | 84 | -TOC | Patient | 52% | 97% | 45% |
| Buchmann et al. [36] | 27 | -TOC | Region | 65.1% | 97.6% | 32.5% |
| Van Binnebeek et al. [41] | 53 | -TOC | Lesion | 60.1% | 99.9% | 39.8% |
| Morgat et al. [43] | 19 | -TOC | Lesion | 20% | 76% | 56% |
| Srirajaskanthan et al. [44] | 51 | -TATE | Lesion | 11.9% | 74.3% | 62.4% |
| Deppen et al. [38] | 78 | -TATE | Patient | 72% | 96% | 24% |
| Sadowski et al. [40] | 131 | -TATE | Lesion | 30.9% | 95.1% | 64.2% |
(n = number of patients; Δ = difference between the sensitivity of 111In-DTPA-octreotide and 68Ga-DOTA-peptide imaging).
The affinity profiles of the 68Ga-DOTA-peptides show some differences (see Table 1), with the most prominent being the fact that 68Ga-DOTANOC also has high affinity for SSTR5 and to a lesser extent for SSTR3, while 68Ga-DOTATOC shows some affinity towards SSTR5 and 68Ga-DOTATATE only binds to SSTR2 [23,35]. On the other hand, the affinity of 68Ga-DOTATATE for SSTR2 is an order of magnitude higher than that of the other 68Ga-DOTA-peptides. Therefore, some differences in lesion detection rate might be expected. A meta-analysis regarding the diagnostic role of 68Ga-DOTATOC and 68Ga-DOTATATE reported a high sensitivity (93% and 96%, respectively) and specificity (85% and 100%, respectively) for both tracers [46]. A head-to-head comparison of 68Ga-DOTATOC and 68Ga-DOTATATE in 40 NET patients showed a comparable diagnostic accuracy, although significantly fewer lesions were detected with the latter (262 vs. 254) [47]. In a similar head-to-head comparison of 68Ga-DOTATATE and 68Ga-DOTANOC in 20 NET patients, a comparable diagnostic accuracy was found as well with both tracers, with a slight, but not statistically significant difference in the number of detected lesions (130 vs. 116) [48]. Conversely, in another comparison study in 18 GEP NET patients, 68Ga-DOTANOC PET detected significantly more lesions than 68Ga-DOTATATE (238 vs. 212 out of 248 lesions), but the authors state that the clinical relevance of this observation has to be confirmed in larger trials [49]. Currently, there is no recommendation on which type of 68Ga-DOTA-peptide is preferred [17,34,50] and logistic reasons such as availability of the precursor peptide will guide the choice in clinical practice. Moreover, there are some practical obstacles to the implementation of 68Ga-DOTA-peptide PET that may differ from country to country. Examples are lack of 68Ge/68Ga generators registered or cleared for human use, no availability of precursor peptide for human use, no reimbursement or the implementation is economically not viable.
Radionuclide
Gallium-68 has the theoretical advantage that it is available from 68Ge/68Ga generators and as such cyclotron independent. However, despite the excellent results achieved with 68Ga-DOTA-peptides, their use in routine clinical practice is often limited to large nuclear medicine departments. Unlike the 99Mo/99mTc generator, the current generation of 68Ge/68Ga generators requires a dedicated radiopharmacy staff. Moreover, production and quality control of gallium-68 radiopharmaceuticals is subject to strict regulations imposed by pharmaceutical legislation [51,52]. Nevertheless, due to the significant boost to clinical PET by the introduction of 68Ga-PSMA-HBED-CC [53], more and more nuclear medicine departments installed these 68Ge/68Ga generators and production facilities. Furthermore, the aforementioned issues may be largely solved with the next generation 68Ge/68Ga generators that have received regulatory approval (e.g. IRE ELiT, Fleurus, Belgium) and the use of kit-based labeling approaches, such as SomaKit TOCTM (Advanced Accelerator Applications S.A.) and NETSPOT® (Advanced Accelerator Applications USA), that have received approval by the European Medicines Agency (EMA) and the Food and Drug Administration (FDA), respectively. As the overall activity yield per production batch is low (capacity of two to four patients per production) and half-life of gallium-68 is relatively short (68 minutes), there is only limited potential for centralized production and distribution. However, some possibilities may be opened up in this field by advances in the cyclotron production of gallium-68 [54].
Another disadvantage of gallium-68 is its relatively high positron energy (Emean = 0.83 MeV) and thus relatively long positron range (Rmean = 3.5 mm), which may compromise spatial resolution [55]. Therefore, the possibilities of using other PET radionuclides for SSTR imaging are currently being explored. The physical characteristics of all radionuclides relevant for PET imaging discussed below are summarized in Table 4.
Table 4
Physical characteristics relevant for PET imaging of the discussed radionuclides, with Emean and Emax the mean and maximum positron energy, respectively, and Rmean and Rmax the mean and maximum positron range calculated in water, respectively
| Isotope | Half-life | Positron branching ratio (%) | Emean (MeV) | Emax (MeV) | Rmean (mm) | Rmax (mm) | Gamma branching ratio (%) | Eγ (MeV) |
|---|---|---|---|---|---|---|---|---|
| Fluorine-18 | 109.8 min | 96.9 | 0.250 | 0.634 | 0.6 | 2.4 | - | - |
| Scandium-44 | 3.97 h | 94.3 | 0.632 | 1.474 | 2.4 | 6.9 | 99.9 | 1.157 |
| Copper-64 | 12.7 h | 17.5 | 0.278 | 0.653 | 0.8 | 2.5 | 0.47 | 1.346 |
| Gallium-68 | 67.8 min | 87.7 | 0.836 | 1.899 | 3.5 | 9.2 | 3.2 | 1.077 |
| 1.2 | 0.353 | 0.822 | 1.1 | 3.4 | ||||
| 88.9 | 0.829 | 3.5 | ||||||
| Terbium-152 | 17.5 h | 8.0 | 1.337 | 2.97 | 6.2 | 15.0 | 63.5 | 0.344 |
| 5.9 | 1.186 | 2.62 | 5.4 | 13.1 | 9.5 | 0.271 | ||
| 20.3 | 1.14 | 5.1 |
For gallium-68 and terbium-152 only the positrons from the two highest positron branching ratios are listed in italics. The total positron branching ratio, Emean and Rmean are listed in bold. Furthermore, the most relevant prompt gammas are given. Data from the Laboratoire National Henri Becquerel (http://www.nucleide.org/Laraweb), Brookhaven National Laboratory (http://www.nndc.bnl.gov/nudat2), and National Institute of Standards and Technology (https://www.nist.gov/pml/radiation-dosimetry-data).
Copper-64
A frequently used non-standard PET radionuclide is copper-64 [54,56]. Its half-life (12.7 hours) allows for centralized production, while its low positron energy (Emean = 0.28 MeV) corresponding to a short positron range (Rmean = 0.8 mm) allows for high spatial resolution PET imaging [54,55]. Copper-64 has a relatively low positron branching ratio of 17.5% and its decay is accompanied by emission of β- particles and Auger electrons adding up to its radiation burden. Therefore, it could be used for therapeutic purposes as well, making it suited for theranostic applications [57], taking into account extensive shielding needed in practice due to the simultaneous high energy gamma and positron emission. Copper-64 can either be produced in a reactor or with a cyclotron [54,57].
As early as 2001, Anderson et al. reported a first clinical evaluation of the dosimetry and pharmacokinetics of 64Cu-TETA-octreotide - copper-64 bound to octreotide through the chelator 1,4,8,11-tetraazacyclotetradecane-N,N’,N’’,N’’’-tetraacetic acid (TETA) (see Figure 1) - in eight NET patients and its diagnostic properties were compared to 111In-DTPA-octreotide [58]. In two patients, 64Cu-TETA-octreotide detected clearly more lesions than 111In-DTPA-octreotide. In one patient mild uptake in a lung lesion was observed with 111In-DTPA-octreotide, but not picked up by 64Cu-TETA-octreotide. However, delayed images which might have shown the lesion were not available for this patient. Pharmacokinetic assessment revealed fast blood clearance with partial urinary excretion. On the other hand, the percentage injected activity in the liver increased with time due to dissociation of the copper-64 isotope, indicating poor in vivo stability [57,58].
The next clinical studies on copper-64 labeled SSAs date from more than 10 years later. Pfeifer et al. prospectively evaluated 64Cu-DOTATATE in a first-in-human study in 14 NET patients [59]. PET images with high spatial resolution were obtained. High and stable tumor-to-background ratios were observed on both the early (one hour post injection (p.i.)) and late (three hours p.i.) PET scans, indicating a high tracer internalization rate. Although some dissociation of copper-64 was suggested by increasing hepatic activity, in vivo stability of the tracer was sufficient for imaging purposes. All patients underwent conventional 111In-DTPA-octreotide SPECT/CT as well. Additional lesions were detected on 64Cu-DOTATATE PET in six patients (43%) and in five of these patients lesions were identified in organs not previously known as disease-involved. No lesions were observed with 111In-DTPA-octreotide SPECT that were not revealed by 64Cu-DOTATATE PET.
In a subsequent larger prospective study, Pfeifer et al. confirmed the diagnostic superiority of 64Cu-DOTATATE PET over 111In-DTPA-octreotide SPECT by means of a head-to-head comparison in 112 NET patients [60]. PET images were acquired one hour after injection. The sensitivity and diagnostic accuracy of 64Cu-DOTATATE PET (both 97%) were significantly higher than those of conventional 111In-DTPA-octreotide SPECT (87% and 88%, respectively). Twice as many lesions were detected using 64Cu-DOTATATE PET, and more importantly, in 40 patients (36%) lesions were identified in organs not previously known as disease-involved. In 35 of these 40 patients the true-positive nature of these supplemental involved organs was confirmed by long-term follow-up of 42-60 months.
Of special interest is a recent study published by Johnbeck et al. comparing 64Cu-DOTATATE and 68Ga-DOTATOC PET in 59 NET patients on a head-to-head basis [61]. On a patient level, 64Cu-DOTATATE and 68Ga-DOTATOC performed equally well. However, 64Cu-DOTATATE detected significantly more additional true-positive lesions, confirmed by at least 30 months of follow-up, than 68Ga-DOTATOC (33 versus 7). The authors attributed this difference in lesion detection rate to the shorter positron range of copper-64, with consequent higher spatial resolution and less partial volume effect, rather than to the use of a different peptide. Figure 4 shows an example of a patient where more lesions are seen in the intestinal region with 64Cu-DOTATATE than with 68Ga-DOTATOC. Tumor-to-background ratios as a measure for image contrast were not significantly different between the two tracers. Although the radiation burden of 64Cu-DOTATATE is higher than that of 68Ga-DOTATOC (5.7-8.9 mSv vs. 2.8-4.6 mSv), the use of 64Cu-DOTATATE offers several advantages for use in routine clinical practices associated to the half-life of copper-64, such as supply to peripheral sites from a central production unit and a more flexible scanning window ranging from one hour to at least three hours after injection. A preclinical evaluation of 64Cu-DOTATOC has been published [62], but to our knowledge no subsequent clinical studies have been performed.

PET/CT (left) and PET (right) scans with 68Ga-DOTATOC and 64Cu-DOTATATE of a patient with intestinal NET and multiple metastases. Additional lesions are seen in the intestinal region with 64Cu-DOTATATE. This research was originally published in JNM. Johnbeck CB, Knigge U, Loft A, Berthelsen AK, Mortensen J, Oturai P, Langer SW, Elema DR and Kjaer A. Head-to-head comparison of 64Cu-DOTATATE and 68Ga-DOTATOC PET/CT: a prospective study of 59 patients with neuroendocrine tumors. J Nucl Med. 2017;58:451-457. © SNMMI.
Copper-64 has also been successfully coupled to TATE by means of a new bifunctional chelator, 5-(8-methyl3,6,10,13,16,19-hexaaza-bicyclo[6.6.6]icosan-1-ylamino)-5-oxopentanoic acid (MeCOSar), forming 64Cu-SARTATE [63]. Initial preclinical results are promising showing a high uptake in SSTR2-positive tumors [63]. Further preclinical and clinical studies on this new radiopharmaceutical are ongoing (Australian New Zealand Clinical Trials Registry (ANZCTR) identifier: ACTRN12615000727549) [57].
Fluorine-18
Among β+-emitting radioisotopes, fluorine-18 is the most commonly used PET radionuclide in clinical practice and offers several logistic and physical advantages over gallium-68. Large amounts of fluorine-18 activity (> 370 GBq) can be produced with a cyclotron and the half-life (109.8 minutes) is long enough to allow transport to remote hospitals without an on-site cyclotron and it is short enough to avoid extended irradiation of patients. Furthermore, it predominantly decays by positron emission (96.9%) with a low positron energy (Emean = 0.25 MeV) leading to a short positron range (Rmean = 0.6 mm) [55].
Meisetschläger et al. evaluated the fluorine-18 labeled SSA, Gluc-Lys-(18F-fluoropropionyl)-Lys-Tyr3-octreotate (Gluc-Lys-[18F]FP-TOCA) (see Figure 1), in 25 patients with SSTR-positive tumors seen on 111In-DTPA-octreotide scan and performed a direct comparison in 16 of these patients [64]. Gluc-Lys-[18F]FP-TOCA showed a fast and high tumor uptake and was rapidly cleared from the blood, mainly through the kidneys. Tumor uptake reached a plateau at about 40 minutes after injection. In contrast to SSAs labeled with radiometals whose fragments remain trapped after cellular internalization, no such trapping has been observed for SSAs-based radiopharmaceuticals labeled with fluorine-18 such as Gluc-Lys-[18F]FP-TOCA [64,65]. Nevertheless, more than twice as many lesions were observed with Gluc-Lys-[18F]FP-TOCA than with 111In-DTPA-octreotide. However, a serious impediment to the implementation of Gluc-Lys-[18F]FP-TOCA in routine clinical practice is its time-consuming multistep synthesis with limited radiochemical yield (20%-30%) [64]. Gluc-Lys-[18F]FP-TOCA has been applied in a few small clinical studies [65-67], but to our knowledge no further large clinical trials have been performed.
18F-fluoroethyl-triazole-Tyr3-octreotate (18F-FET-βAG-TOCA) (see Figure 1) represents an alternative 18F-octreotate radioligand with a more practical and shorter synthesis route and reasonable radiochemical yield [68]. Following the promising results in preclinical models [69], Dubash et al. carried out a first-in-human study in nine NET patients evaluating the biodistribution and dosimetry of 18F-FET-βAG-TOCA [70]. The tracer showed a rapid blood clearance with both renal and biliary elimination, whereas 68Ga-DOTA-peptides are mainly eliminated through the kidneys. As such, the highest absorbed dose was received by the gallbladder. Overall, the dosimetry of 18F-FET-βAG-TOCA was similar to other fluorine-18 labeled tracers. Tumor-to-background ratios were high and comparable to values reported for 68Ga-DOTA-peptide PET, resulting in images with excellent contrast. Larger clinical trials for this promising tracer, including a direct comparison with 68Ga-DOTATATE PET/CT are currently ongoing [70].
Another fluorine-18 based SSA that is subject of recently initiated clinical trials in NET patients is Al18F-1,4,7-triazacyclononane-1,4,7-triacetate-octreotide (Al18F-NOTA-octreotide) (see Figure 1) (clinicaltrials.gov identifier: NCT03511768). The Al18F-labeling method developed by McBride et al. combines the advantages of a chelator-based radiolabeling method with the unique properties of the radionuclide of choice, fluorine-18 [71]. In this method, fluorine is firmly bound to Al3+ forming [18F]AlF which is then complexed by a suitable chelator, conjugated to a vector molecule of interest [72]. Al18F-NOTA-octreotide was developed by Laverman et al. and has been compared to 111In-DTPA-octreotide and 68Ga-NOTA-octreotide in preclinical models [73,74]. Al18F-NOTA-octreotide proved to have the highest in vitro binding affinity for the SSTR (see Table 2), while a biodistribution in AR42J tumor-bearing mice showed that both tumor uptake and pharmacokinetics were similar with an excellent in vitro and in vivo stability.
Other promising fluorine-18 based tracers for SSTR imaging identified in preclinical studies include 18F-silicon-fluoride-acceptor (SiFA) and 18F-SiFAlin octreotate derivatives [75-77], 18F-trifluoroborate octreotate (18F-AMBF3-TATE) [78] and 18F-fluoroglycosylated octreotate (18F-FGlc-TATE) [79].
Scandium-44
Scandium-44 has more recently emerged as a promising radionuclide for PET imaging. There are several methods to produce the radionuclide, for instance by means of a 44Ti/44Sc generator, or using a cyclotron allowing to produce higher quantities [54,80,81]. Scandium-44 mainly decays through positron emission (94.3%) with a somewhat lower positron energy than gallium-68 (Emean = 0.63 MeV) and accordingly lower positron range (Rmean = 2.4 mm). Its half-life of 3.97 hours is convenient for centralized production and distribution [81]. Even more promising is its use in theranostic applications. Although very similar to gallium(III)-68, the chemical behavior of scandium(III)-44 even more closely resembles that of the therapeutic radiometals, such as lutetium-177 and yttrium-90 [82]. Therefore, scandium-44 may represent an attractive alternative to gallium-68 for imaging and dosimetry prior to lutetium-177 based therapy [82]. However, the need for pre-therapy dosimetry has diminished due to the favorable results of the NETTER-1 trial. Moreover, since the half-life of scandium-44 is still limited compared to lutetium-177 (3.97 hours vs. 6.65 days) evaluation at time points later than three days is not possible. With the isotope scandium-47 (100% β- emission), scandium-44 potentially also possesses a true therapeutic match, although research in this area is still in its infancy [83,84].
Several preclinical studies have been published on various scandium labeled SSAs, such as 44Sc-DOTATOC [80,85], natSc-DOTATATE [86], 44Sc-DOTANOC [81,87] and 44Sc-1,4,7-triazacyclononane, 1-glutaric acid-4,7-acetic acid(NODAGA)-NOC [87].
Rösch et al. performed a clinical proof-of-principle study using generator-produced 44Sc-DOTATOC [80,88]. High quality PET images of a patient with SSTR-positive liver metastases were acquired at early time points and up to 18 hours after injection. Singh et al. published a proof-of-concept study using cyclotron-produced 44Sc-DOTATOC in two patients with metastatic NET [89]. Interestingly, scandium-44 was produced at the cyclotron facility at the Paul Sherrer Institut (PSI) in Switzerland and subsequently shipped over 600 km to Zentralklinik Bad Berka (ZBB) in Germany, requiring two and a half half-lives. Eight PET/CT scans were performed at various time points up to 23.5 hours after injection. Excellent tumor uptake was observed with increasing tumor-to-background values over the first four hours after injection. Further clinical studies in larger patient cohorts are scheduled.
Terbium-152
In light of theranostic applications, terbium gained the interest of researchers due to its four medically relevant radioisotopes: terbium-152 and terbium-155 for PET and SPECT imaging, respectively, and terbium-161 and terbium-149 for β- and α-therapy, respectively [84,90]. The latter also offers the possibility of PET imaging, as demonstrated in a preclinical study evaluating 149Tb-DOTANOC in AR42J tumor-bearing mice [91]. Terbium-152, suited for diagnostic PET imaging, has a half-life of 17.5 hours and a positron branching ratio of 20.3% with a relatively high positron energy (Emean = 1.14 MeV) and thus higher positron range (Rmean = 5.1) than gallium-68. Terbium-152 - as well as terbium-149 and -155 - can be produced by high-energy proton-induced spallation in tantalum foil targets [90]. Just like lutetium-177, it belongs to the group of radiolanthanides. Therefore, it can be stably coupled to the chelator DOTA and used for radiolabeling of SSAs [84]. In a preclinical study in AR42J tumor-bearing mice, the biodistribution of 152Tb-DOTANOC was found to be in good agreement with that of 177Lu-DOTANOC [92]. These findings suggest that terbium-152 could serve as a theranostic agent for lutetium-177 based therapy. A clinical proof-of-concept was published by Baum et al. in 2017 using 152Tb-DOTATOC in a patient with well-differentiated metastatic NET of the terminal ileum [93]. Terbium-152 was produced at the Isotope mass Separator On-Line (ISOLDE) facility in CERN in Switzerland, shipped to PSI for separation from the collection matrix and quality control and finally transported to ZBB in Germany for radiolabeling. PET/CT images were acquired at various time points up to 24 hours after injection. All known tumor lesions, visualized on a previous 68Ga-DOTATOC PET scan, were clearly identified. Images were noisier, compared to this previous gallium-68 based PET. This was attributed to the lack of prompt γ-correction by the PET software. Even at 24 hours after injection, increased uptake was observed in several metastases. The authors concluded that the longer half-life of terbium-152, as compared to gallium-68, enabling imaging at later time-points, makes terbium-152 particularly valuable for dosimetry prior to radionuclide therapy [93]. However, the production of terbium-152 is challenging and currently imposes an important constraint on its implementation in routine clinical practice.
Vector
Developments in SSTR PET radiopharmaceuticals do not only focus on the choice of radionuclide but also on the characteristics of the vector molecule. The radiopharmaceuticals described above contain the somatostatin receptor agonist octreotide, or an analog of octreotide, as vector molecule, with a predominance of TOC and TATE. Improved tumor targeting may be achieved for instance by using vector molecules having a higher binding affinity for the SSTR or a broader affinity profile for the different receptor subtypes or by using compounds recognizing a higher number of binding sites, such as the SSTR antagonists.
Somatostatin receptor agonists
Many tumor types that show SSTR expression, predominantly express SSTR2 [12], which makes it the main target for the development of SSTR ligands for imaging and therapy. However, as mentioned above, a wide variability in subtype expression has been observed across and within different tumor types [12]. Therefore, there is great interest in SSTR ligands with a broader affinity profile to increase tumor uptake and to expand the number of tumors eligible for SSTR imaging [94]. Of the 68Ga-DOTA-peptides currently used in clinical practice, 68Ga-DOTANOC has the widest affinity profile with high affinities for SSTR2, SSTR5 and to a lesser extent SSTR3 [23]. However, as discussed above, there is no conclusive evidence to prefer this ligand in clinical practice and large trials are warranted [49], but properly powered prospective trials testing this hypothesis will probably not be available for a long time. Another SSA that has been evaluated early on in clinical trials is the long-acting SSA lanreotide (see Figure 1). Coupled to the chelator DOTA, DOTA-lanreotide (DOTALAN) and the radiolabeled compounds 111In/90Y-DOTALAN showed a high affinity for SSTR subtype 2-5 [95]. However, this was only confirmed for SSTR2 and 5 by Reubi et al. [23]. Following promising clinical results with 111In-DOTALAN [96,97], DOTALAN was labeled with gallium-68 to allow PET imaging. After an initial positive evaluation of 68Ga-DOTALAN in 11 patients with lung cancer (three small cell and three non-small cell lung cancer) or thyroid cancer (two medullary and three radioiodine negative thyroid cancer) [98], the tracer was used to identify patients who might benefit from PRRT with 90Y-DOTALAN in a group of NET patients not qualified for PRRT with 68Ga-DOTATOC despite progressive disease [99]. Tumor-to-background ratios were significantly higher for 68Ga-DOTATOC and more tumor sites (106 vs. 53) were detected with 68Ga-DOTATOC than with 68Ga-DOTALAN. Demirci et al. compared 68Ga-DOTALAN with 68Ga-DOTATATE in a group of 11 NET patients and one patient with meningioma. 68Ga-DOTATATE performed better than 68Ga-DOTALAN with a significantly higher lesion uptake and higher lesion detection rate (63 vs. 23 out of a total of 67 tumor lesions detected with both tracers) [100]. Traub-Weidinger et al. evaluated the SSTR status in a heterogeneous group of thyroid cancer patients with progressive disease using two tracers with a distinct SSTR subtype affinity profile, 68Ga-DOTALAN and 68Ga-DOTATOC, in 28 patients [101]. On a patient basis, 12 patients were negative with both SSTR tracers, while mixed results were observed in three patients (two patients negative with 68Ga-DOTALAN, but positive with 68Ga-DOTATOC and one patient vice versa). On a region-based analysis half of the 38 regions positive on SSTR imaging (out of a total of 196 regions) showed mixed results. The authors concluded that, due to the heterogeneous SSTR profile of thyroid cancer lesions, patients with progressive disease may benefit from imaging with different SSTR PET tracers for individualized targeted therapy stratification.
In a first attempt to develop an actual pansomatostatin radiopharmaceutical with high affinity for all SSTR subtypes, the cyclooctapeptide KE108 with pansomatostatin characteristics was modified to couple the chelator DOTA (DOTA-D-Dab-Arg-Phe-Phe-D-Trp-Lys-Thr-Phe or KE88) and allow subsequent radiolabeling with indium-111 and gallium-68 [102]. Although the resulting tracers were able to bind all SSTR subtypes with high affinity, in vitro internalization of the ligand-receptor complex for the SSTR2 was low compared to the SSTR3 and this was reflected by a low in vivo uptake observed in SSTR2-expressing tumors and fast wash out [102]. In another preclinical study, Fani et al. synthesized and evaluated several bicyclic somatostatin-based analogs of which DOTA-Tyr-cyclo(DAB-Arg-cyclo(Cys-Phe-D-Trp-Lys-Thr-Cys)) or AM3, showing a high affinity for SSTR2, SSTR3 and SSTR5, was the most promising [103]. Efficient background clearance and high tumor uptake with 68Ga-AM3 were observed in SSTR2 tumor-bearing mice. Tatsi et al. used the native peptide hormone somatostatin-14 (SS14) as a basis to develop two SS14-derived analogs with a high affinity for all SSTR subtypes and label them with indium-111 [104]. However, both tracers showed poor in vivo stability. Similarly, Maina et al. used the more stable native peptide somatostatin-28 (SS28) as a basis for the development of DOTA-Ser,Leu,D-Trp,Tyr-SS28 (DOTA-LTT-SS28) and the radioligand 111In-DOTA-LTT-SS28 [105]. The compounds displayed a high affinity for all SSTR subtypes and triggered internalization of SSTR2, SSTR3 and SSTR5. 111In-DOTA-LTT-SS28 showed a high and specific uptake in SSTR2-, SSTR3- and SSTR5-expressing xenografts in mice and a much higher in vivo stability than the SS14-derived tracers developed by Tatsi et al. The authors concluded that 111In-DOTA-LTT-SS28 is the first true (pan)somatostatin radioligand and may serve as a model for the further development of pansomatostatin radioligands [105]. Very recently, Liu et al. reported the development of gallium-68 labeled pasireotide (SOM230 or PA1), a longacting synthetic SSA with a high affinity for SSTR1, SSTR2, SSTR3 and SSTR5 [106,107]. The resulting radiotracer 68Ga-DOTA-PA1 displayed a significantly higher in vitro uptake than 68Ga-DOTATATE in three human lung cancer cell lines: lung adenocarcinoma (A549), lung squamous carcinoma (H520) and pulmonary giant cell carcinoma (PG). PET images of A549 tumor-bearing mice showed a high tumor uptake and better signal-to-noise ratio with 68Ga-DOTA-PA1 than with 68Ga-DOTATATE and the PET signal correlated with the total expression of SSTRs and not only SSTR2, as determined by Western blotting. The authors concluded that 68Ga-DOTA-PA1 and its analogs may hold potential for SSTR imaging in clinical practice, especially lung tumors [107].
Therapeutic purposes have also driven some new developments in the vector part of SSTR ligands aiming for higher tumor retention to improve therapeutic efficiency and preferably lower kidney dose to reduce PRRT toxicity. A recent example is given by Tian et al. who conjugated an Evans blue (EB) analog onto octreotate which allows reversible binding of EB-TATE to albumin to prolong half-life in blood, resulting in a SSA with long circulation time [108]. EB-TATE was then labeled with yttrium-90 through the chelator DOTA and injected in AR42J tumor bearing mice. 90Y-DOTA-EB-TATE showed high tumor uptake resulting in a complete regression of the tumors [108]. These promising results were quickly translated into a first-in-human clinical trial by Zhang et al. evaluating the safety, pharmacokinetics and dosimetry of 177Lu-DOTA-EB-TATE in five NET patients as compared to 177Lu-DOTATATE in three other NET patients [109]. The new compound was well tolerated without adverse symptoms. Tumor dose was 7.9-fold higher with 177Lu-DOTA-EB-TATE, while the effective dose was not significantly different between 177Lu-DOTA-EB-TATE and 177Lu-DOTATATE. However, kidney and bone marrow dose increased 3.2- and 18.2-fold, respectively [109], so this radiopharmaceutical does not offer an improved therapeutic ratio compared to the current radiopharmaceutical of choice, 177Lu-DOTATATE.
Somatostatin receptor antagonists
All SSTR-targeting radiopharmaceuticals described above are somatostatin agonists. After binding to the SSTR, the ligand-receptor complex is usually internalized, allowing tracer metabolites to accumulate in the target cells [110]. For a long time, it was believed that this process of internalization and subsequent accumulation of the radioligands was essential for high-contrast imaging of SSTR-positive lesions [111]. However, in 2006, Ginj et al. observed in cell cultures expressing human SSTR2 and SSTR3, that antagonists labeled considerably more receptor sites than agonists [110]. This was reflected by the significantly higher tumor uptake seen in mice bearing SSTR2 and SSTR3-expressing tumors after injection with the corresponding antagonist as compared to those injected with the agonist [110]. Especially striking was the fact that counterintuitive to this observation the SSTR2 antagonist, 111In-DOTA-pNO2-Phe-c(D-Cys-Tyr-D-Trp-Lys-Thr-Cys)D-TyrNH2 (111In-DOTA-BASS) (see Figure 1), showed a more than sevenfold lower affinity for the SSTR2 than the SSTR2 agonist 111In-DTPA-TATE in this study (50% inhibitory concentration (IC50) of 9.4 ± 0.4 nM vs. 1.3 ± 0.2 nM) [110]. In 2011, Wild et al. published a pilot study in five patients, one metastatic follicular thyroid carcinoma and four NETs, evaluating the biodistribution, tumor uptake and detection of tumor lesions with the SSTR antagonist 111In-DOTA-BASS in a head-to-head comparison with 111In-DTPA-octreotide [112]. Tumor uptake was up to four times higher with the antagonist, while renal, liver and spleen uptake were lower. 111In-DOTA-BASS detected more lesions than 111In-DTPA-octreotide (25 vs. 17 out of 28). The three missed bone lesions were negative on the 111In-DTPA-octreotide scan as well.
Further advances in the preclinical setting in search of more potent SSTR2 antagonists labeled with PET radioisotopes led to the identification of Cpa-c[D-Cys-Aph(Hor)D-Aph(Cbm)-Lys-Thr-Cys]-D-Tyr-NH2 (JR11) (see Figure 1) as a promising compound for implementation in the clinical field [113]. Based on the results of previous affinity studies and an in vivo biodistribution study [114], 68Ga-NODAGA-JR11 (68Ga-OPS202) was selected for a first clinical evaluation by Nicolas et al. [115,116]. 12 patients with GEP NET and a positive 68Ga-DOTATOC PET/CT scan in the previous six months were administered two microdoses of 68Ga-NODAGA-JR11 with ascending peptide masses at different study visits and underwent subsequent PET/CT scans. 68Ga-NODAGA-JR11 showed a fast blood clearance and favorable biodistribution with both peptide doses as compared to 68Ga-DOTATOC (lower hepatic, pancreatic, gastro-intestinal and splenic uptake with 68Ga-NODAGA-JR11) [115,116]. This was reflected by higher tumor-to-background ratios observed with 68Ga-NODAGA-JR11 and further translates to a significantly higher number of detected tumor lesions and higher lesion-based overall sensitivity (94% and 88% for 50 µg and 15 µg 68Ga-NODAGA-JR11, respectively, vs. 59% for 68Ga-DOTATOC) [116]. An example is shown in Figure 5. The effective dose is in line with 68Ga-DOTA-peptides (24 ± 2 µSv/MBq for 68Ga-NODAGA-JR11 vs. 21 ± 3 µSv/MBq for 68Ga-DOTATATE and 68Ga-DOTATOC [117]), although some differences in organ doses are observed due to a slightly different biodistribution [115]. Overall, 68Ga-NODAGA-JR11 was well tolerated [115]. However, SSTR antagonists could possibly counteract the effects of SSAs, which may be important in patients with functioning NETs [111]. Therefore, caution is required in anticipation of more safety data [111]. Currently a multicenter clinical trial evaluating the optimal dose and safety of 68Ga-NODAGA-JR11 for PET imaging is ongoing (clinicaltrials.gov identifier: NCT03220217).

Maximal-intensity projections (A and C) and PET/CT (B and D) scans with 68Ga-NODAGA-JR11 (A and B) and 68Ga-DOTATOC of a patient with ileal NET and bilobar liver metastases. Liver magnetic resonance imaging (MRI) was performed four months later, with delayed post-contrast acquisitions (E) and diffusion-weighted images (F), confirming the additional metastases missed or questionable (arrow with question mark) with 68Ga-DOTATOC. Note the lower background activity in the liver, intestine and thyroid with 68Ga-NODAGA-JR11. Dashed lines in (A and C) denote the level of transversal slices in (B, D, E and F). This research was originally published in JNM. Nicolas GP, Schreiter N, Kaul F, Uiters J, Bouterfa H, Kaufmann J, Erlanger TE, Cathomas R, Christ E, Fani M and Wild D. Sensitivity comparison of 68Ga-OPS202 and 68Ga-DOTATOC PET/CT in patients with gastroenteropancreatic neuroendocrine tumors: a prospective phase II imaging study. J Nucl Med. 2018;59:915-921. © SNMMI.
The higher tumor uptake achieved with SSTR antagonists may also prove useful for therapeutic purposes. Preclinical studies comparing 177Lu-DOTA-JR11 (177Lu-OPS201) with 177Lu-DOTATATE observed higher tumor uptake and longer residence times, resulting in higher tumor doses delivered by the antagonist as compared to the agonist [118,119]. In a clinical pilot study by Wild et al. in four patients with progressive NETs, more than threefold higher tumor doses and twofold higher tumor-to-kidney and tumor-to-bone marrow dose ratios were observed using a test dose of 177Lu-DOTA-JR11 as compared to 177Lu-DOTATATE [120]. All patients were subsequently treated with 177Lu-DOTA-JR11, resulting in partial remission in two patients, mixed response in one patient and stable disease in the last patient, and as such proving the clinical feasibility of PRRT using radiolabeled SSTR antagonists. Currently, a multicenter clinical trial evaluating the safety and efficacy of 177Lu-DOTA-JR11 (clinicaltrials.gov identifier: NCT02592707) as well as a study evaluating the theranostic couple 68Ga-DOTA-JR11 and 177Lu-DOTA-JR11 (clinicaltrials.gov identifier: NCT02609737) are ongoing.
Radiolabeled SSTR antagonists might also prove to be especially useful for imaging and therapy of cancer types with a typically lower SSTR expression such as breast cancer [121]. Several preclinical studies observed enhanced tumor targeting in various human SSTR2-expressing tumor samples, including breast carcinoma, by means of in vitro autoradiography using an SSTR2 antagonist in comparison to the SSTR2 agonist [121-123]. This finding was not confirmed in a recent preclinical study by Dude et al. using the human luminal breast cancer model, ZR-75-1, with endogenous SSTR2 expression and negligible expression of other SSTR subtypes, where 68Ga-NODAGA-JR11 had a lower tumor uptake than 68Ga-DOTATOC and 68Ga-DOTATATE [124]. This was tentatively explained by the authors by the fact that they used an endogenously expressing cell line, which may have a lower amount of low-affinity, antagonist-specific binding sites. Interestingly, although 68Ga-DOTATATE has a higher affinity for the SSTR2 than 68Ga-DOTATOC, the latter was found to have the highest tumor uptake. Additional studies are warranted to further investigate the role of SSTR2 antagonists in breast cancer imaging [124].
Conclusion
Advances in SSTR PET ligands occur on two major fronts: the radionuclide and the peptide vector. Other radionuclides could offer a solution to practical, economical and regulatory barriers to the adoption of 68Ga-DOTA-peptide PET, with additional physical advantages such as lower positron range and longer half-life. Developments concerning peptide vectors are mainly driven by the need for improved lesion targeting, especially for tumors with low SSTR expression. Therefore, advances on both fronts are largely complementary. Several promising new PET ligands for clinical SSTR imaging are currently in the pipeline and good results have been demonstrated in phase II trials. Clinical adoption in the near future is a realistic scenario.
Acknowledgements
This research was funded by the project from “Kom op tegen Kanker”: “PET/MR imaging of the norepinephrine transporter and somatostatin receptor in neural crest and neuroendocrine tumors for better radionuclide therapy selection” and received support from Research Foundation-Flanders (FWO) (G0D8817N). Frederik Cleeren is a Postdoctoral Fellow of FWO (12R3119N). Christophe M. Deroose is a Senior Clinical Investigator at the FWO.
Disclosure of conflict of interest
Christophe M. Deroose received grants and personal fees from Novartis, Terumo, AAA, Ipsen, Sirtex, Bayer outside the submitted work.
The information comes from:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6261874/