Abstract
Alpha particle-emitting isotopes are being investigated in radioimmunotherapeutic applications because of their unparalleled cytotoxicity when targeted to cancer and their relative lack of toxicity towards untargeted normal tissue. Actinium-225 has been developed into potent targeting drug constructs and is in clinical use against acute myelogenous leukemia. The key properties of the alpha particles generated by 225Ac are the following: i) limited range in tissue of a few cell diameters; ii) high linear energy transfer leading to dense radiation damage along each alpha track; iii) a 10 day half-life; and iv) four net alpha particles emitted per decay. Targeting 225Ac-drug constructs have potential in the treatment of cancer.
Introduction
The treatment of cancer with targeted radionuclide therapy is a maturing field that has achieved significant success with the introduction of two FDA approved drugs adding new modalities to the current cancer therapy arsenal of surgery, chemotherapy and external beam radiation. The specificity imparted by the targeting vehicle (e.g., antibodies (IgGs), peptides, small molecules) can be complemented by attaching a cytotoxic payload to yield a potent therapeutic effect. Particle-emitting radionuclides are some of the most promising cytotoxic moieties when linked to tumor-targeting carrier molecules. To date, much of the work has been done with beta particle-emitting isotopes. Iodine-131 Tositumomab (Bexxar) and Yttrium–90 Ibritumomab Tiuxetan (90Y-Zevalin) are FDA approved beta particle-emitting IgGs used to treat B-cell non-Hodgkin’s lymphoma [1].
The increased availability and improved radiochemistry of alpha particle-emitting nuclides for targeted therapy have presented novel possibilities for their use in radioimmunotherapy (RIT). Alpha particles offer key advantages over beta particles, in particular are the high linear energy transfer (LET) and the limited range in tissue. The high alpha particle LET is on the order of 100 keV/μm and it can produce substantially more lethal double strand DNA breaks per alpha track than beta particles when traversing a cell nucleus. The alpha particle tracks are relatively short and thus have a limited range in tissue (on the order of a few cell diameters). This confines the toxic effect to a relatively small field - within a few cell diameters from the site of decay versus the much longer-ranged beta particles. 90Y for example, has a maximum range on the order of several hundred cell diameters and thus deposits energy in the tumor as well as the surrounding normal tissue. The number of particle track transversals through a tumor cell nucleus that was necessary to kill the cell was considerably lower for alpha particles than for beta particles and it has been estimated that one alpha particle transversal can kill a cell [2]. This higher biological effectiveness seems nearly independent of oxygen concentration, dose rate and cell cycle position.
Preclinical research has demonstrated the potential of alpha particle-emitting isotopes in RIT [3,4]. Alpha emitting nuclides displayed cytotoxicity in a model of leukaemia that was resistant to beta- and gamma-radiotherapy and doxorubicin chemotherapy [5]. There are a number of alpha particle-emitting nuclides considered for application in targeted therapy displaying half-lives ranging from minutes to days. 213Bi was one alpha particle-emitting nuclide (t1/2 = 46 min) that has been proposed for therapeutic use and has been evaluated clinically. However, 213Bi is generator-produced and has a very short half-life. The clinical use of 213Bi presents the logistical dilemma of eluting the generator, radiolabeling the targeting molecule, administering a dose, and allowing sufficient time for targeting. All of these steps consume valuable time that decreases the effective dose administered. An innovative alternative to 213Bi was the use of its parent nuclide, 225Ac, which has a ten-day half-life and 4 net alpha particle-emissions per decay. In vitro cytotoxicity data using the same antibodies labeled with either 213Bi or 225Ac demonstrated that several logs less 225Ac radioactivity was necessary to reach LD50, presumably because of the multiple alpha emissions and the 300-fold longer half-life [6]. The enhanced potency of 225Ac versus 213Bi was also directly demonstrated in a murine model of human prostate cancer [6,7]. This article reviews the literature of 225Ac dealing with its production and supply, physical, chemical and biological properties, dosimetry, and clinical use as a radiotherapeutic agent in cancer therapy.
225Ac production and supply
Actinium was aptly named for the Greek aktis or aktinos, meaning ray or beam [8]. The element was discovered by Debierne in 1899 and Giesel in 1902 [9]. Actinium occurs naturally in association with uranium radionuclides and 225Ac can be obtained either from the decay of 233U or from the neutron transmutation of 226Ra by successive n,γ capture decay reactions via 227Ac, 228Th to 229Th [10–12]. Currently, there are two sources of 225Ac that have been used in clinical trials: (1) the U.S. Department of Energy, Oak Ridge National Laboratory (ORNL) in Oak Ridge, TN, United States of America and (2) the Institute for Transuranium Elements in Karlsruhe, Germany. The 225Ac at both sites was derived from 233U that was produced as a component of the U. S. molten salt breeder reactor program [13–15], and had been in long-term storage at ORNL. The bulk of this high purity and low specific activity 229Th was separated from waste material associated with the original production of the 233U. This 229Th yields 225Ac that was produced as a “carrier-free” nuclide and was suitable for use in clinical research applications. The 225Ac from both sources has been used to construct 213Bi producing generators [16,17] for Phase I and I/II clinical treatment of leukemia [18] and the 225Ac from Oak Ridge used to directly radiolabel an antibody for application in a Phase I clinical trial treating leukemia (ongoing clinical trial [19]).
An 225Ac generator based on a design that adsorbed 229Th oxide onto a titanium phosphate resin was described by Geerlings et al. [20]. Elution of this 229Th cow with dilute nitric acid yielded a mixture of radionuclides: 225Ac, 225Ra, and 224Ra. Another downstream column, containing Dowex 50 WX8, was used to purify the 225Ac by removing the 225Ra, 224Ra, and the 224Ra decay products. In 1993, it was proposed that the 225Ac thus produced could be used to label an antibody or be affixed to a resin as a parent for a 213Bi generator product.
The separation method used at Oak Ridge National Laboratory to isolate 225Ac from the 229Th stock allows 229Th, 225Ra, and 225Ac to reach equilibrium (45 d) and then carrier-free 225Ra and 225Ac were separated from the thorium stock in nitric acid using anion exchange chromatography [15]. Concentration of the 225Ra/225Ac eluate was effected by evaporation or neutralization and co-precipitation. Recently, this group has described a four-step chemical separation procedure, employing both anion and cation exchange chromatography, to process their current supply of 150 mCi of 229Th into 225Ac [14]. Over an 8-week period, approximately 100 mCi of 225Ac was yielded per processing campaign and the product shipped in 5–6 batches. Following the initial process run of a campaign yielding ~ 50–60 mCi 225Ac, the radium pool was reprocessed bi-weekly to yield the additional 225Ac shipments. This material is currently being utilized in both a 213Bi- and an 225Ac-labeled antibody trial at Memorial Sloan-Kettering Cancer Center (MSKCC) [18]. The average radionuclidic purity was 99.6% ± 0.7% 225Ac with ≤ 0.6% 225Ra contaminant and an average 229Th content of 4 (+5/−4) × 10−5 %.
The separation and purification method to yield 225Ac from a 229Th source that is currently employed at the Institute for Transuranium Elements entails a 229Th stock is batch loaded in nitric acid onto 0.5 L of Dowex anion exchange resin [13]. This process utilizes a combination of extraction and ion exchange chromatographic methods to obtain carrier-free, clinical quality 225Ac with > 95% overall yield. Based upon their stock of 215 mg of 229Th, they can isolate 43 mCi of 225Ra and 39 mCi of 225Ac every 9 weeks. This 225Ac has been used in clinical generators [17] to produce 213Bi for radiolabeled radioimmunotherapeutic (RIT) pharmaceuticals at MSKCC [17] and in collaborations with a number of other sites [13].
A liquid 229Th/225Ac generator used a process that entails maintaining a stock 229Th solution in an ammonium citrate solution in order to eliminate the radiolysis and degradation experienced with solid sorbents [21]. As the 225Ra and 225Ac reach equilibrium with the 229Th, they are isolated in a one-step cation exchange process. The 229Th breakthrough was effectively removed in a single separation cycle by changing the pH of the solution. This process takes advantage of the differences in the stability constants of thorium (K1 = 1013, K2 = 108) and actinium (K1 ~ K2 = 106) citrate complexes [22].
Cyclotron production is an alternate strategy for 225Ac production that employs proton irradiation of 226Ra leading to 225Ac via [p,2n] reactions [10,11,23]. Theoretically, irradiation of 1 mg of 226Ra should yield 35 mCi of 225Ac [11]. Recently, the feasibility of cyclotron produced 225Ac was demonstrated and maximum yields were reached with an incident proton energy of 16.8 MeV [23] using the 226Ra(p,2n)225Ac reaction. In this work, 0.0125 mg of 226Ra yielded 0.0021 mCi 225Ac after irradiation of a 36 mm2 target with a 10 μA proton current for 7 h. No significant differences were found in the radionuclidic purity of the cyclotron product when compared to 225Ac produced via the 229Th method [13] and 213Bi produced from this 225Ac was found to label antibody constructs with approximately 90% yield.
Chemical and radionuclide properties of 225Ac
225Ac decay (see Figure 1) yields six principal radionuclide progeny in the decay cascade to stable 209Bi [3]. A single 225Ac (t1/2 = 10.0 d; 6 MeV α particle) decay yields net 4 alpha and 3 beta disintegrations, most of high energy and 2 useful gamma emissions of which the 213Bi 440 keV γ emission has been used in imaging drug distribution [17]. These daughters are 221Fr (t1/2 = 4.8 m; 6 MeV α particle and 218 keV γ emission), 217At (t1/2 = 32.3 ms; 7 MeV α particle), 213Bi (t1/2 = 45.6 m; 6 MeV α particle, 444 keV β− particle and 440 keV γ emission), 213Po (t1/2 = 4.2 μs; 8 MeV α particle), 209Tl (t1/2 = 2.2 m; 659 keV β− particle), 209Pb (t1/2 = 3.25 h; 198 keV β− particle) and 209Bi (stable). Given the 10.0 d half-life of 225Ac, the large alpha particle emission energies, and the favorable rapid decay chain to stable 209Bi this radionuclide was recognized as a potential candidate for use in cancer therapy [19]. Figure 1 illustrates the decay scheme of 225Ac.

The potential for using 225Ac as a therapeutic radionuclide was limited many years by the paucity of suitable chelating moieties capable of stably binding this radionuclide as well as controlling the fate of the daughters [21]. Additionally, the chemistry of actinium was not explored or well developed. Diamond and Seaborg studied the elution profiles of the transuranium elements in hydrochloric acid on cation-exchange resin and concluded that the actinides may form complex ions with chloride to a greater extent than the lanthanide elements based on the partial covalent character of the actinide bonds involved in the hybridization of the 5f orbitals [24]. The actinium(III) ionic radius was reported as 0.111 nm [25]. Radiopolarographic reduction of the 225Ac(III) ion in aqueous solution in the presence of 1,4,7,13,16-hexaoxacyclooctadecane (18-CROWN-6) and suggested the formation of a divalent actinium cation [26]. In the absence of 18-CROWN-6, the measured E1/2 value was −2.15 V versus SCE and as increasing concentrations of 18-CROWN-6 were added, the E1/2 value shifted to a more negative potentials in a linear fashion. They concluded from this study that the Ac(II) ionic radius was 0.125 nm and the electronic configuration was [Rn]6d1. The overall hydrolytic constant, β3, for the hydrolysis of 225Ac(III) in aqueous NaClO4 (μ = 0.1) solution was determined using the electromigration method [26]. A plot of the 225Ac ion velocity as a function of pH showed a constant velocity of 5.4 × 10−4 cm2/Vs in the pH interval from 4–10, indicating that no hydrolytic process occurred until pH = 10. At pH 10, the velocity dropped steeply and by pH 11 the velocity was 0. A value of pβ3 = 31.9 ± 0.2 was calculated for the hydrolytic reaction, Ac3+ + 3H2O → Ac(OH)3 + 3H+, where β3 was the hydrolytic constant. When the pH was less than 4, a 10–15% decrease in ion mobility was measured.
Attempts to utilize 225Ac as a tumoricidal agent steered the evaluation of a different complexing agents and chelates in order to enhance tumor uptake and avoid normal organ uptake. 225Ac complex stability improved by trial and error and a trend was recognized as one moved from simple complexing agents to acyclic chelates to macrocyclic chelates. Two related macrocyclic chelates, in particular, were identified as potentially useful and further explored as moieties to attach to targeting monoclonal antibody carriers. The first was 1,4,7,10,13,16-hexaazacyclohexadecane-N,N′,N″,N‴,N′‴,N″‴-hexaacetic acid (HEHA) and the second was 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA). These chelates are related because they are both macrocycles that present carboxylic acid and amine functionalities to the metal-ion, albeit with different denticity, macrocycle size, and overall charge. As will be described below, the 225Ac complex with HEHA demonstrated less stability than the 225Ac complex with DOTA in vivo in the experiments described. In addition, the two monoclonal antibody/antigen systems that were examined using HEHA-constructs were non-internalizing immune complexes and the targeted constructs could still release the daughters systemically. The 225Ac released from the HEHA was distributed to liver and bone and the subsequent release of its daughters contributed to acute radiotoxicity as did the daughters from the targeted, but not internalized parent. The 225Ac complex formed with DOTA was considerably more stable in vivo and the antibodies selected for these studies formed internalizing immune complexes with their respective antigen targets.
Isothiocyanate-functionalized-DOTA derivatives were selected as the most promising to pursue for coupling to antibody molecules from out of a group of potential 225Ac chelate compounds: diethylenetriaminepentaacetic acid (DTPA), 1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetraacetic acid (TETA), DOTA, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrapropionic acid (DOTPA), 1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetrapropionic acid (TETPA), and 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetramethylenephosphonic acid (DOTMP). The bifunctional chelating agents MeO-DOTA-NCS, (α-(5-isothiocyanato-2-methoxyphenyl)-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid and 2B-DOTA-NCS, 2-(p-isothiocyanatobenzyl)-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid were both evaluated and compared as their respective [225Ac]DOTA-antibody construct. A two-step labeling method was developed using several different IgG systems [27]. The chelation reaction yield in the first step was 93% ± 8% radiochemically pure. The second step, which couples the [225Ac]DOTA-SCN moiety to the IgG, yielded constructs that were 95% ± 5% radiochemically pure and with mean percent immunoreactivity ranging from 25–81%, depending on the antibody used but consistent for each IgG system. This methodology met the requirement for high temperature labeling of the DOTA chelate with 225Ac without sacrificing the biological activity of the protein.
Biological studies in animal models
Free metal-ion, complexing and chelating agent studies
The biodistribution of a mixed 225Ac, 169Yb and 148Pm sodium citrate solution (pH 6.5) was examined in normal rats and mice bearing adenocarcinoma tumor implants [28]. The rats were injected i.v. while the mice were injected intraperitoneally (i.p.). Each animal received 40 kBq (1080 nCi) of 225Ac in a citrate solution. Animals were euthanized 5 h post-injection and blood, liver, femur, urine, and tumor were harvested. Samples were measured 1 d after harvest after secular equilibrium was established. As expected for a rapidly clearing small molecular weight metal-citrate complex, the percent injected dose (%ID) of 225Ac per gram of blood was low, 0.06 and 1.2 for rats and mice, respectively. Inter-species liver and femur values were different; the rats had 5.7 and 1.2 %ID 225Ac/g while the mice had 38.7 and 16.8 %ID 225Ac/g, respectively. Tumor tissue in mice accumulated 3.5 %ID 225Ac/g.
The influence of varying ethylenediaminetetramethylenephosphonic acid (EDTMP) solution concentrations on the biodistribution of 225Ac was determined in tumor-bearing mice [29]. Swiss nude mice were implanted with subcutaneous (s.c.) T380 human colon carcinoma and injected i.v. via the tail vein with approximately 50 kBq 225Ac (1350 nCi) in formulations varying the concentration (0, 0.01, 0.05, 0.1, 0.2, 0.5, 1, 2, 10 and 30 mM) of EDTMP or in a 1 mM citrate control solution, adjusted to pH 6.5. Animals were sacrificed at 15 h and tissues were harvested and measured using a high resolution Ge-spectrometer after secular equilibrium was established. [225Ac]citrate control and [225Ac]EDTMP solutions (up to 0.1 mM EDTMP) demonstrated high liver uptake (40 %ID/g). The higher EDTMP concentrations exhibited less than 1 %ID 225Ac/g accumulated in the liver, suggesting that the excess EDTMP assisted the clearance of the 225Ac.
The biodistribution, dosimetry and radiotoxicity of 225Ac complexed with acetate, ethylenediaminetetraacetic acid (EDTA), 1,4,7,10,13-pentaazacyclopentanadecane-N,N′,N″,N‴, N′‴-pentaacetic acid (PEPA), and the A″ isomer of N-[(R)-2-amino-3-(4-nitrophenyl)propyl]-trans-(S,S-cyclohexane-1,2-diamine-N,N,N′,N″,N″-pentaacetic acid (CHX-A″-DTPA) was examined in female BALB/c mice [30]. Animals were sacrificed at 1h, 4h, 24h, 5d, and 8d following i.v. tail vein injection of 92 kBq (2500 nCi) of each of the 225Ac complexes. Tissue samples were harvested, held for 4 h, and then counted using a NaI(Tl) γ-scintillation counter. Data expressed as the %ID/g again demonstrated that the liver was the major site of 225Ac localization for all four small molecule complexes studied. Liver accumulation increases according to the decreasing strength of the 225Ac-complex: CHX-DTPA ~ PEPA > EDTA > acetate. For example, the 24h liver biodistribution data values were approximately 13, 15, 53, and 110 %ID/g for the respective [225Ac]CHX-DTPA, [225Ac]PEPA, [225Ac]EDTA, 225Ac-acetate complexes. When the data were expressed as the % injected dose per organ it was shown that for [225Ac]CHX-DTPA, the bone was the predominant localization site and the liver next (at 24 h, 42 %ID/bone and 13 %ID/liver). Liver and femur accumulation presumably resulted from the loss of 225Ac from the chelators. Absorbed dose values for 225Ac were estimated based upon the data from [225Ac]CHX-DTPA and [225Ac]EDTA. Values are reported as Gy per 92 kBq of injected dose of 225Ac-complex. For [225Ac]CHX-DTPA and [225Ac]EDTA the doses to liver were 30.4 and 117.8; 7.8 and 15.4 to bone; and 3.2 and 2.1 to kidney, respectively. Both acute and chronic toxicity were assessed by organ system damage and white blood cell (WBC) counts as a function of the dose administered. Two mice receiving 92, 185, 370, 740 kBq (2,500, 5,000, 10,000, 20,000 nCi) of [225Ac]CHX-DTPA were sacrificed at 2, 5, 7, 53 d post-injection (the latter two time-points were included for animals that had not succumbed to radiation toxicity during the study). The control was i.v. injected CHX-DTPA chelate-alone in MES buffer. Tissues from these animals were harvested, H&E stained and fixed, evaluated histopathologically, and graded for radiation damage. The tissues that showed the most radiation-induced toxicity were the bone marrow, spleen, gastrointestinal tract, and the liver. At the 92 kBq dose level, the WBC, spleen and bone marrow were rated as having loss of cellular numbers, integrity, orientation, or structure. At the 185 kBq dose level the WBC, spleen, bone marrow, liver, GI tract, and kidney all were rated as having loss of cellular numbers, integrity, orientation, or structure and evidence of cellular necrosis [30].
Another series of 225Ac-labeled chelate complexes was prepared and their biodistribution measured in normal BALB/c mice [31]. One of these complexes was reported to exhibit improved in vivo stability relative to the others in the series examined. The chelates studied were EDTA, CHX-A-DTPA, PEPA, DOTA, HEHA, and acetate. 92.5 kBq of each complex (2500 nCi) in MES buffer at pH 6.2 was injected into normal female BALB/c mice via the tail vein. Biodistributions were performed at 1, 4, 24, 120 h and samples were counted after 4 h to allow for secular equilibrium. All of the complexes rapidly cleared the blood with < 2 %ID/g in 1 h. The order of most 225Ac distributed into tissue to the least was acetate > EDTA > CHX-A″-DTPA ~ PEPA > DOTA > HEHA. Consistent with the studies described above, the loss of 225Ac from an acyclic chelate was greatest and reflected in the high uptake in liver and bone and poor whole body clearance. [225Ac]HEHA was rapidly excreted within 1 h and only 0.17 % ID/g remained (approximately 100 nCi of the 2500 nCi injected).
Targeting monoclonal antibodies
The development of tumor specific agents was addressed by preparing antibody constructs with the various chelate candidate molecules identified in the previously described reports. The cytotoxicity of an [225Ac]DTPA-antibody construct was reported in vitro using a murine IgG1 that targets a carbohydrate structure associated with the EGF receptor expressed on the human epidermoid A431 tumor cell line [32]. The specific antibody construct was more potent than the non-specific control construct. However, the DTPA chelate moiety clearly did not stably bind the 225Ac in these experiments as demonstrated by the 225Ac constructs being capable of killing target cells only slightly better than similarly labeled 213Bi-antibodies. In another radiolabeled antibody-DTPA construct study, the biodistribution of [225Ac]DTPA-201B, which targeted murine lung endothelial thrombomodulin, was examined [31]. The construct efficiently targeted the lung but the 225Ac had a very short tissue t1/2 of 4–5 h as compared with the [125I]-201B construct with a t1/2 of 4–5 d. Obviously, the DTPA was unable to stably bind the 225Ac in vivo and this chelate was not going to advance the use of 225Ac in RIT applications.
HEHA is a multidentate, macrocyclic chelate having 6 carboxylic acids and 6 amino nitrogens (compare with PEPA having 5 of each and DOTA having 4 of each). Given the larger macrocycle size, greater number of coordinating ligands, and overall negative charge, the [225Ac]HEHA-antibody complex was hypothesized to be a potentially useful RIT construct. The properties of HEHA were useful for rapidly clearing the simple complex in vivo, but did not address the potential stability should the HEHA chelate be used in an IgG construct that would presumably have a longer biological half-life [31]. The interest in the HEHA chelate led to the description of the synthesis of an isothiocyanate bifunctional chelate (BFC) derivative and the subsequent conjugation to three different antibodies, and radiolabeling of with 225Ac [33]. One of the [225Ac]HEHA-IgG constructs was evaluated for serum stability in fetal bovine serum at 37°C over a 3 d period. After 0.4 h of incubation, 99% of the 225Ac was still associated with the construct, but at 1, 3, 5, 24, and 48 h this value decreased rapidly to 77, 73, 69, < 50, and < 50%, respectively. It was determined that the HEHA rapidly released the 225Ac associated with the targeting antibody and bound to serum proteins.
The [225Ac]HEHA-201B antibody construct was evaluated for vascular targeted therapy of lung tumors in the first reported 225Ac RIT study of in vivo [34]. This study performed biodistribution, dosimetry and therapeutic efficacy studies in female BALB/c mice with the EMT-6 mammary carcinoma as the model. At 1 and 4 h post-injection, 300 %ID/g of 225Ac was distributed to the lung tissue, however, the 225Ac cleared the lung with a t1/2 of 49 h and the released 225Ac accumulated predominantly in the liver, spleen, and bone. It was calculated that a dose of 6 Gy per μCi was delivered to the lungs and about three-fold less to other tissues. A RIT study was performed where 18.5 kBq of construct administered per mouse and 10% of the tumor bearing animals survived 23 d vs. 11 to 15 d for untreated controls. 80% of the animals treated with 37 kBq of 225Ac drug had the tumor eradicated, but died at 16 d from acute radiotoxic effects (total bone marrow ablation, splenic atrophy, damage to the lining of the stomach and intestine). In subsequent studies, no therapeutic window could be identified that would effectively treat tumor but spare the host. The HEHA chelate released the 225Ac and the non-internalizing antibody-antigen complex compromised the use of this system for RIT.
It was hypothesized that if an antibody armed with 225Ac did not form an internalizing antibody-antigen complex, then a smaller domain-deleted fragment of that antibody could better extravasate and penetrate the tumor where the progeny would remain localized as compared to the native, full-sized IgG [35]. The CC49 antibody and the humanized domain-deleted product, ΔCH2CC49, were converted to HEHA-appended constructs, radiolabeled with 225Ac, and evaluated for biodistribution, microdistribution, and therapeutic efficacy in mouse models with s.c. and/or intramuscular (i.m.) LS174T xenografts. The biodistribution data revealed that the [225Ac]HEHA-CC49, [225Ac]HEHA-ΔCH2CC49, and [225Ac]HEHA-control antibody constructs accumulated after 24 h with 25, 18, and 10 %ID/g in the s.c. tumors and 8, 9, and 5 %ID/g of in the i.m. tumors, respectively. Liver and spleen accumulated 225Ac, which increased over a week, presumably due to release from the HEHA chelate. The retention of the progeny was investigated by calculating the ratio of 213Bi to 225Ac in the tumors over an 8 d period and it was found that there was little difference between the CC49 and the domain-deleted fragment. In one RIT study, 800 nCi of [225Ac]HEHA-CC49, [225Ac]HEHA-ΔCH2CC49, or [225Ac]HEHA-control were administered to SCID/LS174T models having both s.c. and i.m. xenografts 9 d post-tumor implant. The maximum tolerated dose (MTD) of the constructs was 800 nCi in these animals and all mice exhibited radiotoxicity by day 6 and were sacrificed on day 8. There was no statistical difference in the tumor sizes in this study based upon treatment. A second RIT study was conducted in a Swiss nude/LS174T model 9 d post tumor implant. Animals had either s.c. or i.m. implants, but not both. Animals with i.m. tumors responded best to treatment with 500 nCi [225Ac]HEHA-CC49, with statistically smaller tumors than those treated with [225Ac]HEHA-ΔCH2CC49, control [225Ac]HEHA-IgG, or cold, unlabeled CC49. Animals with s.c. tumors all responded to treatment with 500 nCi [225Ac]HEHA-CC49, [225Ac]HEHA-ΔCH2CC49, or control [225Ac]HEHA-IgG, but the latter two groups suffered from radiotoxicity. A third RIT study focused on the Swiss nude/LS174T model treated 6 d post tumor implant with 0, 250, or 500 nCi of [225Ac]HEHA-ΔCH2CC49 or 500 nCi of the [225Ac]HEHA-IgG control. Animals had either s.c. or i.m. implant, but not both. There were no statistical differences in the tumor sizes per group in the s.c. implant groups. It was concluded that only marginal therapeutic effect could be attained with the [225Ac]HEHA-CC49 and no therapeutic effect was observed using the domain-deleted fragment, contrary to the penetration/retention hypothesis. RIT with HEHA constructs was limited by radiotoxicity to normal organs.
The Targeting Nanogenerator approach
Controlling the fate of the parent radionuclide and the ensuing progeny would be the key to harnessing the therapeutic potential of 225Ac. The initial step was to identify a suitable chelating agent that would yield stable 225Ac complexes in vivo and thereby shape the pharmacokinetic profile of the parent nuclide by keeping it associated with the targeting carrier immunoglobulin for a long time. Managing the distribution, metabolism and clearance of the daughters was a more daunting task. As described above, the attempts which focused on developing a single chelate moiety to accommodate the parent and the progeny, proved too difficult given the range of different periodic properties of these daughters and using antibody fragments did not enhance tumor penetration and or retention. Previous workers had concluded that 225Ac-antibody constructs were too unstable and that the progeny presented an untenable pharmacological problem. The nanogenerator system altered this paradigm and clearly demonstrated the ability to safely and efficaciously use 225Ac as an extraordinarily potent tumor-selective molecular sized generator in both established solid carcinomas or disseminated cancers.
The approach which was taken that focused on first, stably chelating the 225Ac for delivery in vivo to a target cell; second, internalizing the 225Ac-antibody construct into the target cell; third, retaining the progeny inside the target cell and harnessing their cytotoxic potentials; and fourth, minimizing the loss of the daughters to non-target tissues and thus minimizing systemic radiotoxicity. This strategy was called the 225Ac atomic nanogenerator. DOTA was found to be a stable chelate for 225Ac and was attached to the antibody using a robust thiourea chemical linkage. The targeting agents were internalizing IgGs which stably transported the 225Ac to the cell which bound and modulated the construct into the cell where the progeny were retained and decayed. This approach proved extremely cytotoxic to the targeted cancer cells and largely eliminated systemic toxicity to the host. The 225Ac delivered to the cancer cell was effectively a therapeutic nanogenerator of multiple alpha particle emissions within the target cell [6].
The first practical application of 225Ac in targeted RIT without any accompanying systemic radiotoxicity utilized the nanogenerator approach [6]. This process was dependent on the stable chelation of the parent 225Ac radionuclide and the efficient delivery and internalization of the construct at the tumor target site. Controlling the [225Ac]DOTA-antibody pharmacokinetics was the key to the use of 225Ac as a therapeutic agent. Further, the progeny retained at the target site harnessed their cytotoxic potentials and contributed to safe and effective tumor therapy. It was discovered that a very stable 225Ac complex with DOTA could be rapidly formed at 60°C. The ensuing [225Ac]DOTA complex could then be coupled to an IgG using the thiourea linkage [27].
The stability in vitro of [225Ac]DOTA-HuM195 was compared to an analogous 177Lu-labeled construct in 100% human serum, 100% mouse serum, and 25% human serum albumin at 37°C for 15 d. The 225Ac construct displayed stability similar to the 177Lu analogue, with less than 5% loss of 225Ac from the chelate over 15 d. The stability results in all three conditions were similar. Stability in vivo was determined using 10 female nude mice injected i.v. with 300 nCi of [225Ac]DOTA-HuM195. The % 225Ac that was bound to the HuM195 in the mouse serum was determined as a function of time. IgG bound 225Ac was determined using a Protein A binding assay, HPLC size exclusion chromatography (SEC) analysis of the serum, and a cell based immunoreactivity assay. The results of the Protein A bead assay at 5 time-points from 2.5 to 120 hours, showed that the mean % 225Ac that was bound to the HuM195 was 98.2 to 99.9%, respectively. HPLC SEC analysis revealed that the 225Ac species in the serum was associated with an antibody sized molecule. The immunoreactivity assay of a serum sample indicated that 63% of the 225Ac species was bound to CD33 expressing AL67 cells versus 3% bound to non-specific Daudi cells. In conclusion, 225Ac bound to HuM195 remained associated with the IgG following injection into a mouse over a 5 d period, demonstrating the stability of the drug in vivo [6].
The in vitro cytotoxicity of 225Ac-antibody constructs that were designed to specifically target HL60 leukemia cells (HuM195 (anti-CD33)); Daudi and Ramos lymphoma cells (B4 (anti-CD19)); MCF7 breast carcinoma cells (trastuzumab (anti-HER2/neu)); LNCaP.FGC prostate carcinoma cells (J591 (anti-PSMA)); and SKOV3 ovarian cancer cells (trastuzumab (anti-HER2/neu)) were examined using very small doses of nanogenerators. The LD50 values of the 225Ac constructs ranged from 0.3 to 74 Bq/mL (0.008 to 2 nCi/mL) and were 2–4 logs lower than activity values for corresponding 213Bi alpha-particle emitting antibodies (see Table I). Controls at low specific activities (target sites were blocked by addition of excess unlabeled ‘cold’ antibody) did not show specific binding of the nanogenerators to target, and were used to evaluate non-specific cytotoxicity. The LD50 values were 10- to 625-fold greater in the blocked controls [6].
Table I
Comparison of the cytotoxicity in vitro of 225Ac- versus 213Bi-antibody constructs.
| Antibody | 225Ac construct ED50 (pCi/mL) | 213Bi construct ED50 (pCi/mL) | Cancer | Cell type |
|---|---|---|---|---|
| HuM195 | 8 | - | AML | HL60 |
| HuM195 | - | 200,000 | AML | HL60 |
| B4 | 60 | - | B-NHL | Daudi |
| B4 | - | 280,000 | B-NHL | Daudi |
| J591 | 90 | - | Prostate Carcinoma | LNCaP |
| J591 | - | 220,000 | Prostate Carcinoma | LNCaP |
| 3F8 | 100 | - | Neuroblastoma | NMB7 |
| Herceptin | 150 | - | Breast Carcinoma | BT-474 |
| Herceptin | 1300 | - | Ovarian Carcinoma | SKOV3-NMP2 |
ED50 was measured by [3H]thymidine uptake assay [6].
A pharmacokinetic analysis of the 225Ac construct and two of its progeny was performed in vivo by injecting 12 kBq of [225Ac]DOTA-J591 or 12 kBq of [225Ac]DOTA-HuM195 (irrelevant control) i.p. in two groups of male athymic nude mice bearing an i.m. LNCaP tumor xenograft. Approximately, 18 and 21 %ID/g of [225Ac]DOTA-J591 was localized in the tumor at 2 and 3 d, respectively. Tumor samples counted 9 min. after sacrifice/harvest, demonstrated that 221Fr was 88% ± 9% and 213Bi was 89% ± 2% of the 225Ac secular equilibrium levels in the tumor. These results indicate the uptake of the parent IgG construct by the tumor and the retention of the progeny at that location [6]. In toxicity experiments, the MTD in naive 20 g mice was 18.5 kBq (500 nCi) [225Ac]DOTA-IgG. Mice injected with 37 kBq (1000 nCi) of [225Ac]IgG died. Based on these studies, RIT doses were selected that were approximately 40% of MTD [6].
Therapeutic efficacy of [225Ac]DOTA-J591 was evaluated in an i.m. LNCaP tumor model in vivo. Serum prostate specific antigen (PSA) was utilized in this xenograft model to follow tumor growth. The experimental groups of animals had mean PSA values of 2–5 ng/mL on 10 and 12 d after implantation of tumor. At the time the [225Ac]DOTA-J591 was administered on day 12 or 15, the tumors were characterized histologically as vascularized and encapsulated nodules each comprised of tens of thousands of cells. Animals were sacrificed when tumor area was ≥ 2.5 cm2. In the first RIT study, mice were treated on day 15 post-tumor implantation and received 7.2 kBq [225Ac]DOTA-J591 in a single nontoxic administration. These animals had significantly improved median survival times relative to mice treated with a similar dose of [225Ac]DOTA-B4 irrelevant control antibody mixed with unlabeled specific J591 (dual control) or untreated controls. There was no significant difference in survival times between the dual control-treated animals and untreated controls. The median survival time of untreated growth controls in this model was 33 d. The mean and median pre-therapy PSA values measured on day 12 were not significantly different between the three groups of mice. However, on days 28 and 42, the PSA values of [225Ac]DOTA-J591 treated animals were significantly lower than the PSA values for the dual control-treated animals and untreated controls. There was no significant difference between the dual control-treated animals and untreated controls at either time. Additionally, no acute radiotoxicity was observed [5].
In a second RIT study, mice were treated on day 12 after LNCaP tumor implantation with a single, non-toxic administration of 7.8 kBq [225Ac]DOTA-J591 which caused tumor regression and significantly improved the median survival times of these mice to 158 d compared to the 63 d in the mice treated on day 15. PSA values decreased from pre-therapy levels in many of the animals following treatment to low and undetectable levels and remained undetected in the 14 of the 39 treated animals which exhibited prolonged survival. These mice survived at least 10 months and had no measureable PSA or evidence of tumor at the time of sacrifice (293 d). Animals treated with unlabeled J591 (0.004 or 0.04 mg) on day 12 post-implantation had no prolongation of median survival (37d and 35d, respectively). The therapeutic efficacy was dependent on antibody specificity, the administration of the 225Ac-generator, and the treatment time after implantation [6].
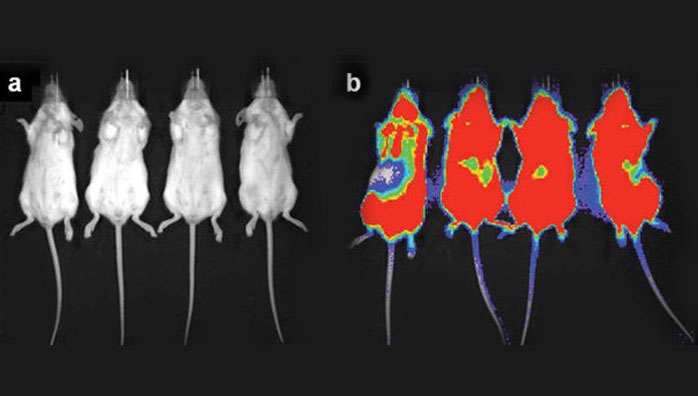
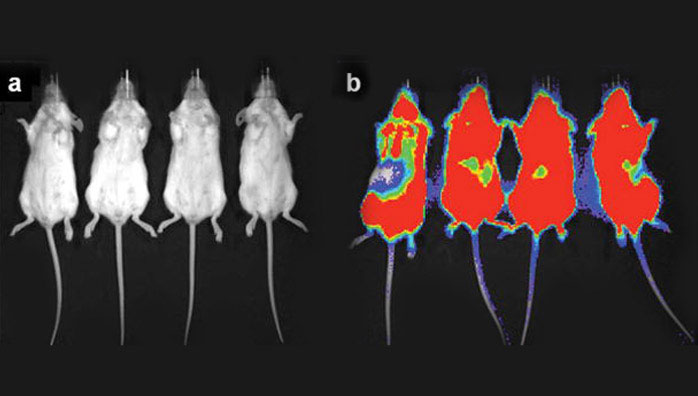
In order to determine if other tumor types could be treated with 225Ac-generator constructs, a disseminated human Daudi lymphoma cell mouse model using [225Ac]DOTA-B4 as the therapeutic agent was investigated. SCID mice were treated 1 d after tumor dissemination with a single administration of specific [225Ac]DOTA-B4 (three different dose levels), irrelevant control [225Ac]DOTA-HuM195 (two dose levels), or unlabeled B4. Control mice receiving the irrelevant [225Ac]DOTA-HuM195 had median survival times from xenograft of 43 d (5.6 kBq) and 36 d (1.9 kBq). Mice receiving 0.003 mg unlabeled B4 per mouse had a median survival time of 57 d. The mice receiving a single injection of [225Ac]DOTA-B4 showed dose-related increases in median survival times 165 (6.3 kBq), 137 (4.3 kBq), and 99 d (2.1 kBq), respectively. This dose response of [225Ac]DOTA-B4 was significant and about 40% of mice treated at the highest dose were tumor-free at 300 d and the experiment concluded on day 310 [6]. The images in Figure 2 demonstrate the potency of the [225Ac]DOTA-B4 drug construct against this disseminated lymphoma model.

Bioluminescence imaging (BLI) of two groups of scid mice that were xenografted with Daudi tumor cells transfected with the green fluorescent protein (GFP) and firefly luciferase (FFLuc) genes [45]. Images were taken on day 17 (a) after treatment with [225Ac]DOTA-B4 or (b) untreated growth controls. In the scid model, the GFP+/FFLuc+ Daudi cells developed into macroscopic, disseminated tumors in the bone marrow and spleen as well as in kidneys, liver, lungs, ovaries, and adipose tissue. BLI clearly showed the presence of lymphoma in the untreated mice while no disease was detected in the mice treated with the [225Ac]DOTA-B4. (n.b., the scale bar indicate the value x 1E6 photons/sec/cm2).
The time of treatment from tumor implantation was examined in the second disseminated lymphoma experiment in vivo. Mice that received treatment on day 1, 3, or 6 post tumor implantation with a single administration of [225Ac]DOTA-B4 (6.3 kBq) had similar prolongation of survival relative to untreated growth controls. Mice that received treatment 13 d after tumor dissemination survived > 165 d. Unlabeled B4 was minimally active in mice with median survival of 44 and 40 d for mice treated with 0.002 mg or 0.20 mg, respectively. Untreated growth controls had a median survival time of 28 d. Therefore, in this lymphoma model, while specificity and dose level were important factors in efficacy, the treatment time after tumor dissemination was less relevant up to a time, at which it was then inversely related to activity. The latter phenomenon may be related to differences in the geometry of the alpha emission eradicating single cells or clusters of tumor cells [6].
Following these initial studies, the same strategy was employed in a RIT experiment in an i.p. mouse model of human SKOV3 ovarian cancer using [225Ac]DOTA-trastuzumab, an anti-HER-2/neu construct [36]. Construct that was administered i.p., had a high tumor uptake, 60 %ID/g at 4 h. Tumor uptake was 3–5-fold higher than liver and spleen. RIT was examined with native trastuzumab and doses of 220, 330 and 450 nCi of [225Ac]DOTA-trastuzumab or [225Ac]DOTA-labeled control antibody at different dosing schedules. Therapy was initiated 9 d after tumor seeding. Groups of untreated control mice and those administered native trastuzumab had median survivals of 33 and 44 d, respectively. Median survival was 52–126 d with [225Ac]DOTA-trastuzumab at various doses and schedules and 48–64 d for [225Ac]DOTA-labeled control IgG. Radiotoxicity occurred with only the highest activity levels administered, but other dose levels were safe. It was concluded that i.p. administration with an internalizing [225Ac]DOTA-labeled anti-HER2/neu antibody significantly extended survival in a mouse model of human ovarian cancer at levels that produce no apparent gross toxicity.
In anticipation of starting human clinical trials, the pharmacokinetics, dosimetry and toxicity of [225Ac]DOTA-HuM195 was investigated in cynomolgus monkeys [37]. The monoclonal antibody, HuM195 (anti-CD33), was the targeting molecule intended for human clinical trials of [225Ac]DOTA-IgG directed against leukemia. In one experiment, two monkeys received a single i.v. dose of the construct at 28 kBq/kg. This dose level was approximately that planned for initial human dose. In another experiment, two animals received a dose escalation schedule of three increasing [225Ac]DOTA-HuM195 doses with a cumulative activity of 377 kBq/kg. Cynomolgus monkeys do not express human CD33 and thus there were no targets for this antibody in this system. The blood half-life of the construct; the ratio of 225Ac:213Bi; the generation of monkey anti-human antibodies (MAHA); haematological indices; serum biochemistries; and clinical observation were the parameters that were measured to evaluate toxicity. Monkeys were euthanized and examined histopathologically when the dose escalation study exhibited toxicity. The blood half-life of [225Ac]DOTA-HuM195 was 12 d and 45% of generated 213Bi daughters were cleared from the blood. MAHA production was not detected. A dose of 28 kBq/kg of 225Ac caused no toxicity at 6 months, whereas a cumulative dose of 377 kBq/kg caused severe toxicity. In the cumulative dosing schedule experiment, single doses of about 37 kBq/kg resulted in no toxicity at six weeks. After 130 kBq/kg was administered, no toxicity was observed for 13 weeks. However, 28 weeks after this second dose administration, mild anemia and increased blood urea nitrogen (BUN) and creatinine were detected indicating renal toxicity. Following administration of an additional 185 kBq/kg, toxicity became clinically apparent. Monkeys were euthanized 13 and 19 weeks after the third dose administration (cumulative dose was 377 kBq/kg). Histopathological evaluation revealed renal tubular damage associated with interstitial fibrosis. In conclusion, the 225Ac nanogenerator constructs may result in renal toxicity and anemia at high doses. The longer blood half-life and the lack of target cell antigens in cynomolgus monkeys may increase toxicity compared to human application. It was concluded that a dose level of 28 kBq/kg could be a safe starting dose in humans, and that hematologic and renal function will require close surveillance during clinical trials.
A model of neuroblastoma meningeal carcinomatosis was treated with an intrathecal (i.t.) administration of [225Ac]DOTA-3F8 construct that targeted ganglioside GD2 [38]. 3F8 is an antibody that specifically binds to ganglioside GD2, overexpressed by many neuroendocrine tumors including neuroblastoma (NB). The [225Ac]DOTA-3F8 construct was prepared and evaluated for radiochemical purity, sterility, immunoreactivity, cytotoxicity in vitro, induction of apoptosis on GD2-positive cells, as well as pharmacologic biodistribution and metabolism of the 225Ac generator and its daughters in a nude mouse xenograft model of NB. Therapeutic efficacy was examined in a nude rat xenograft model of meningeal carcinomatosis and an additional toxicity study in cynomolgus monkeys was performed after i.t. administration. [225Ac]DOTA-3F8 biodistribution in mice showed specific targeting of a s.c. tumor with some redistribution of the 225Ac daughter nuclides mainly from blood to kidneys and to small intestine. In an aggressive meningeal carcinomatosis xenograft nude rat model, i.t. RIT improved survival time two-fold. Increasing the construct specific activity (S.A.) to > 1 MBq/mg improved the therapeutic efficacy relative to lower S.A. preparations. Monkeys injected i.t. with multiple doses of the [225Ac]DOTA-3F8 prepared under clinical manufacturing conditions, did not show any signs of toxicity based on blood chemistry and by complete blood counts or by clinical examination.
Breast cancer spheroids with different HER2/neu expression levels were treated with [225Ac]DOTA-trastuzumab [39]. The breast carcinoma cell lines MCF7, MDA-MB-361, and BT-474 with relative HER2/neu expression of 1:4:18 (determined by flow cytometry) were used. Spheroids of these cell lines were incubated with different concentrations of construct, and spheroid growth was measured by light microscopy over a 50 d period. The activity concentration required to yield a 50% reduction in spheroid volume at 35 d was 18.1, 1.9, and 0.6 kBq/mL (490, 52, 14 nCi/mL) for MCF7, MDA, and BT-474 spheroids, respectively. MCF7 spheroids continued growing, but with a 20–30 d growth delay at 18.5 kBq/mL. MDA-MB-361 spheroid growth was delayed by 30–40 d at 3.7 kBq/mL and at 18.5 kBq/mL, 12 of 12 spheroids disaggregated after 70 d and cells remaining from each spheroid failed to form further colonies. Eight of 10 BT-474 spheroids failed to regrow at a construct concentration of 1.85 kBq/mL. All of the BT-474 spheroids at activity concentrations 3.7 kBq/mL failed to regrow and form colonies. The radiosensitivity of these three cell lines evaluated as spheroids was described as the activity concentration required too reduce the treated-to-untreated spheroid volume ratio to 0.37, denoted DVR37. The external beam radiosensitivity for spheroids of all three cell lines was found to be 2 Gy. After α-particle irradiation using the construct yielded a DVR37 of 1.5, 3.0, and 2.0 kBq/mL for MCF7, MDA-MB-361, and BT-474, respectively.
Targeting the aberrantly formed and angiogenic neovasculature in tumors with an 225Ac-labeled antibody construct has proved very to be effective in prolonging survival and improving subsequent chemotherapy in prostate [40] and adenocarcinoma [41] xenograft models. Angiogenic vascular endothelium (VE) expresses the monomeric cadherin (VE-cadhm) epitope on the cell surface that upon dimerizing with another monomeric copy of VE-cadhm on an adjoining cell forms a tight adherens junction between the cells. The antibody E4G10 binds only to VE-cadhm and not the homodimeric form (the binding region is masked in the homodimer), thus conferring specificity for targeting angiogenic and poorly joined VE cells in vivo while not binding to normal VE or tumor. We have demonstrated that [225Ac]DOTA-E4G10 could specifically irradiate carcinoma VE cells as well as bone marrow-derived endothelial progenitors (42) and delay tumor growth. We have also examined [225Ac]DOTA-E4G10 in vascular targeted strategies to treat animal models of glioblastoma multiforme with similar results (preliminary data). Treatment with 1.85 kBq (50 nCi) [225Ac]DOTA-E4G10 on day 3, 5, 7 and 10 after xenotransplant of LNCaP prostate carcinoma cells achieved highly significant inhibition of tumor growth and lower PSA values 22 d after tumor implantation over [225Ac]DOTA-non-specific IgG and vehicle [40]. The lack of binding to tumor cells and to normal vasculature was demonstrated by flow cytometry, by SPECT imaging and by biodistribution studies. Additionally, subsequent bi-weekly administration of paclitaxel for two weeks resulted in further enhancement of the anti-tumor response to survival times of 182 d, compared to [225Ac]DOTA-E4G10 monotherapy (113 d) and to combination of 225Ac-labeled unspecific IgG with paclitaxel (84 d). The authors concluded that targeting the neovasculature with alpha particles was an effective approach to cancer therapy and that sequential therapy with chemotherapy could potentially result in a synergistic effect when temporal administration was carefully planned.
Disrupting and damaging the vascular endothelial architecture associated with tumor tissue is a viable therapeutic strategy. The endothelial vessels in tumor vasculature often do not exhibit the same hierarchy as in normal tissue. Instead, tumor vascular networks are tortuous and have abnormal component and structural composition. Endothelial cells in these tumors are inefficiently joined with holes, gaps and defects; pericytes are loosely associated with vessels or absent; and basement membranes are inefficiently applied relative to typical normal tissues. Alpha particles are charged helium nuclei that travel approximately 50–80 μm, the dimensions of a typical vessel within a tumor. In addition, individual α-particles are able to kill a target cell due to their deposition of 5–8 MeV in an ionizing track that is several cell diameters in length. Alpha particles are very potent cytotoxic agents in this close vicinity but will largely spare normal tissue; it is this characteristic that offers clear advantages to other known forms of targeted radiation (i.e., β−- or auger-emitters) or stereotactic external beam therapy as a means of effecting selective cell kill.
In the colon adenocarcinoma model system, a novel mechanism of achieving vascular normalization and improved chemotherapy with a cytotoxic anti-vascular agent was described [41]. Selective cytotoxicity was directed towards the tumor neovasculature using short-ranged alpha particles targeted to VE-cadhm. Immunofluorescence and immunohistochemical studies showed that the alpha-targeted vasculature of tumors was largely depleted, and that the remaining vessels appeared more mature as substantiated by accompanying morphological changes and increased pericyte density and coverage. Tumor accumulation and micro-distribution studies with radioactive and fluorescent small molecule drugs showed better uptake and more homogenous distribution of the drugs within [225Ac]DOTA-E4G10 treated tumors (versus controls), thus explaining the enhanced therapeutic response. The results showed not only that 225Ac treatment lead to ablation and remodeling of the tumor vasculature, but also suggested that the resulting vessel normalization improved tumor delivery of small molecules.
A metastatic breast cancer model was investigated and compared the therapeutic efficacy 225Ac-, 213Bi-, and 90Y-labeled anti-rat HER-2/neu monoclonal antibody (7.16.4) as the therapeutic agents. A single 400 nCi administration of [225Ac]-7.16.4 completely eradicated breast cancer lung micrometastases in 67% of HER-2/neu transgenic mice and resulted in long-term survival of these mice for up to 1 y. Treatment with 225Ac-7.16.4 was significantly more effective than 120 μCi of [213Bi]-7.16.4 (median survival, 61 d) and 120 μCi of [90Y]-7.16.4 (median survival, 50 d) as well as untreated control (median survival, 41 d). Dosimetric analysis of the 225Ac-treated mice demonstrated that the metastases received a total dose of 9.6 Gy, significantly greater than the 2.0 Gy from 213Bi or 2.4 Gy from 90Y. Biodistribution studies revealed that 225Ac progeny accumulated in kidneys and probably contributed to the long-term renal toxicity observed in surviving mice. These data suggested that an 225Ac-labeled anti-HER-2/neu monoclonal antibody construct could prolong survival in HER-2/neu-positive metastatic breast cancer patients [43].
Targeting peptides and small molecules
A property of such small molecules that limits their application was that after the dose was administered, a relatively high distribution volume was quickly achieved. This occurs in a time that was similar to the radioactive decay of these nuclides. Therefore, the rapid elimination of these small molecules was not advantageous when used with short half-lived radionuclides. However, 225Ac might be an interesting candidate for small molecules in applications where a high tumor accumulation was possible. Peptide receptor radionuclide therapy (PRRT) was evaluated preclinically with 225Ac-labeled 1,4,7,10-tetra-azacylododecane N,N′,N″,N‴-J-tetraacetic acid-Tyr3-octreotide (DOTATOC) against pancreatic neuroendocrine tumor in a xenograft model [44]. Doses of [225Ac]-DOTATOC up to 20 kBq (540 nCi) were not toxic in mice, while activities greater than 30 kBq (811 nCi) induced tubular necrosis. Biodistribution studies revealed that [225Ac]-DOTATOC effectively localized in the neuroendocrine tumors with some kidney accumulation. Doses of 12–20 kBq (324–540 nCi) of [225Ac]-DOTATOC effectively controlled neuroendocrine tumor growth and showed improved efficacy compared with [177Lu]-DOTATOC and DOTATOC controls.
Carbon nanotube constructs
Innovations in pharmaceutical design are envisioned that seek to improve the potency and specificity of conventional agents by integrating nanomaterials into the drug construct blueprint. For example, novel synthetic nanostructures based on molecules consisting of biologics, radionuclides and carbon nanotubes will have emergent anti-cancer properties because of amplified targeting, binding, imaging, therapeutic and novel pharmacokinetic characteristics. These nanomaterials should therefore exhibit improved potency, specificity, and efficacy relative to conventional agents. Soluble carbon nanotube constructs, containing multiple copies of covalently attached antibodies, and chelated radiometals, have been shown to target lymphoma and adenocarcinoma in imaging and therapeutic studies in animal models of human disease [45,46]. Prototypes of these nanoconstructs rapidly cleared the blood (t1/2 < 1 h), had minimal accumulation in liver, spleen and kidney, and were rapidly cleared intact into the urine (t1/2 ~ 6 min.) by glomerular filtration [47,48].
Metallofullerene carriers
The production and radio-chromatographic properties of a metallofullerene encapsulated 225Ac were reported. Chromatographic results suggested a 3+ oxidation state of Ac in the Ac@C82 complex and the presence of metallofullerene isomers with properties similar to La@C82 [49,50].
Liposomal carriers
Pegylated phosphatidylcholine-cholesterol liposomes with encapsulated 225Ac (passively entrapped) were developed to retain the potentially toxic daughters at the tumor site. More than two 225Ac atoms were successfully entrapped per liposome and more than 88% of the activity was retained over 30 d. The size of the liposomal structures required to contain the daughters makes this approach ideally suited for locoregional therapy (e.g., i.p., intrahepatic artery, or intrathecal) [51].
Improved daughter retention was realized using multivesicular liposomes (MUVEL). MUVELs are large pegylated liposomes with the 225Ac entrapped within smaller lipid-vesicles. This strategy provides confinement of entrapped 225Ac within the region of the liposomal core, away from the outer liposomal membrane. These MUVELs yielded 98% retention of 225Ac and 18% retention of the last daughter 213Bi for 30 d. MUVELs were then conjugated to trastuzumab and exhibited robust binding and internalization by ovarian carcinoma cells. [225Ac]-MUVELs were administered i.p. to animals with disseminated disease and significant tumor uptake of 225Ac and its daughters was detected [52].
Targeting cancer cells that express low levels of antigens is an issue with strategies using radiolabeled carriers with low specific activities. Antibodies may not deliver enough α-emitters to the targeted cancer cells to result in killing, but liposomes with conjugated with targeting antibodies were loaded with high levels of 225Ac to overcome the limitations of low specific activity. As expected these were demonstrated to be therapeutically useful against tumor cells having a low antigen density [53].
Large (approximately 600 nm in diameter) pegylated liposomes conjugated with trastuzumab resulted in swift, specific binding to cancer cells in vitro, followed by cellular internalization. After i.p. administration, these liposomes again exhibited rapid, specific binding to tumors. The large liposomes were cleared slowly from the i.p. cavity, exhibited an increased uptake by the spleen relative to the liver, and specifically targeted tumor. The findings suggested that large targeted liposomes administered i.p. could be a potent drug-delivery strategy for locoregional therapy of i.p. micrometastatic tumors [54].
Imaging
The extreme potency of 225Ac necessitates the small doses administered for preclinical and clinical therapeutic studies; as a consequence, planar SPECT imaging of 225Ac or its 213Bi daughter is not feasible [55]. The development of a novel optical imaging technique was recently reported the used the inherent optical emissions from radionuclide decay for Cerenkov luminescence imaging (CLI) of tumors in vivo. The results correlated with those obtained from concomitant immuno-PET studies using analogous β+-emitting immunoconstructs. Phantom studies confirmed that Cerenkov radiation was observed from a range of positron-, beta-, and alpha-emitting radionuclides using standard optical imaging devices. The change in light emission intensity versus time linearly correlated with radionuclide decay, activity concentration, and the measured PET signal (%ID/g). The value of CLI lies in its ability to image radionuclides that do not emit either β+ or γ-rays and are unsuitable for use with current nuclear imaging modalities. Optical imaging of Cerenkov radiation emission shows excellent promise as a potential new imaging modality for the rapid, high-throughput screening of radiopharmaceuticals [56].
Pharmacokinetic control of the parent and progeny radionuclides and pharmacological interventions
Further efforts to understand the pharmacokinetics of the 225Ac progeny and the elucidation of methods to control their biodistribution and elimination have been described in order to have a better idea of the consequences of therapy in vivo [57]. The combination of stable DOTA chelation of 225Ac, efficient targeting of cell specific epitopes, and internalization of the constructs led to a potent and effective therapeutic nanogenerator strategy no systemic toxicity observed. The internalization of the 225Ac-labeled construct coupled with the stable DOTA chelation during targeting proved useful in harnessing the cytotoxic potential of the daughters and mitigating their effects.
Groups of naïve mice were administered (i.v.) 500 nCi of [225Ac]DOTA-IgG. Metallic progeny chelation was effected with either 2,3-dimercapto-1-propanesulfonic acid (DMPS) or meso-2,3-dimercaptosuccinic acid (DMSA). Both chelating agents significantly reduced the renal uptake of 213Bi; however, DMPS was more effective than DMSA. Diuresis with furosemide or chlorothiazide (CTZ) treatment significantly reduced the renal 213Bi and 221Fr activities. The combination of DMPS with either CTZ or furosemide further reduced renal 213Bi activity. Competitive antagonism with ’cold’ bismuth subnitrate only moderately reduced the renal uptake of 213Bi. In studies with tumor-bearing mice, the tumor ‘sink’ significantly prevented the renal 213Bi accumulation as the daughter was presumably in the tumor. The tumor sink-effect combined with DMPS treatment further reduced renal 213Bi activity. The results indicated that metal chelation, diuresis with furosemide or CTZ, and competitive metal blockade could serve as adjuvant therapies to modify the potential nephrotoxicity of 225Ac progeny. Furthermore, internalization of the parent construct to tumor decreased non-specific organ uptake of 213Bi [57].
A pharmacokinetic study of the short-lived daughter nuclide 221Fr was performed in naïve mice [58]. The majority of the progeny biodistribution studies (described above) focused on 213Bi, so a source of 221Fr was developed for comparison. An 225Ac/221Fr generator was designed and constructed. Briefly, a DOTA-biotin construct [59] was labeled with 225Ac and reacted with an immobilized avidin column. The generator was eluted with normal sterile saline yielding predominantly 221Fr activity. The 221Fr biodistribution study only harvested and counted blood and kidneys because of the short nuclide half-life. The %ID/g of 221Fr was 52.3 ± 8.4 and 5.4 ± 0.3 in the kidneys and blood of these animals (n = 3), respectively.
Radiobiological consequences of non-specific internal radiation from the progeny and pharmacological interventions
Some of the systemically released 225Ac progeny accumulated in the kidneys. The ensuing renal tubulointerstital changes were investigated in order to elucidate the radiobiological effects of the daughters in the kidney [60]. Toxicological and histopathological evaluations of nonhuman primates (cynomolgus monkeys) which received a cumulative activity of 377 kBq/kg in a dose escalation schedule of [225Ac]DOTA-HuM195, revealed mainly renal tubular damage associated with interstitial fibrosis [37]. Upto this time, the mechanism of radiation nephropathy that resulted from targeted radionuclide therapies was poorly understood.
Naïve mice were administered 350 nCi of [225Ac]DOTA-HuM195 and the subsequent functional and morphological changes in their kidneys were assessed longitudinally. Renal irradiation from free, radioactive 225Ac progeny led to time-dependant reduction in renal function manifested as tissue pallor and an increased blood urea nitrogen titer. Corresponding histopathological changes were observed in the kidneys. Glomerular and tubular cell nuclear pleomorphism, karyorrhexis, tubular cell injury and lysis were observed as early as 10 weeks. Progressive thinning of the cortex due to widespread tubulolysis, collapsed tubules, glomerular crowding, decrease in glomerular cellularity and interstitial inflammation and an elevated juxtaglomerular index were noted at 5–7 weeks post-treatment. By 35 – 40 weeks, regeneration of simplified tubules with tubular atrophy and loss and focal interstitial fibrosis had occurred. A lower juxtaglomerular cell index with focal cytoplasmic vacuolization was observed and suggested increased degranulation. Increased tubular and interstitial TGF-β1expression and a corresponding increase in the extracellular matrix deposition was noticed only at 40 weeks post-injection. These findings suggest that internally delivered α-particle radiation-induced loss of tubular epithelial cells and triggered a procession of adaptive changes that resulted in progressive morphological damage accompanied by a loss of renal function.
Radiation nephropathy that followed internal alpha particle irradiation of kidneys was ameliorated by pharmacologically modifying the functional and morphological changes in mouse kidneys following injection of [225Ac]DOTA-HuM195 using several different agents [61]. The 350 nCi dosage yielded a 27.6 Gy dose to the kidneys. Mice were randomized to receive captopril (ACE inhibitor), L-158,809 (Angiotensin II receptor-1 blocker), spironolactone (aldosterone receptor antagonist) or a placebo control. Forty weeks after [225Ac]DOTA-HuM195 injection, placebo-control mice showed significant increase in BUN, dilated Bowman spaces and tubulolysis with basement membrane thickening. Spironolactone treatment significantly prevented the development of adverse histopathological and functional changes vs. placebo controls. The Angiotensin II receptor-1 blocker offered moderate protection. Captopril treatment accentuated the functional and histopathological damage. In conclusion, low-dose spironolactone, and to a lesser extent, angiotensin receptor-1 blockade, offered renal protection in a mouse model of internal alpha particle irradiation.
Dosimetry
Actinium-225 and its progeny present unique radiobiological and dosimetric challenges to the medical physicist because of the multiple progeny, the diverse progeny chemical characteristics and pharmacokinetic profiles, and the short-range, high LET α-particle emissions. Recently, MIRD Pamphlet No. 22 was published to address these issues for 225Ac and other α-particle emitters [62,63]. A plan was developed for estimating absorbed dose to organs following the administration of radionuclides with multiple progeny in order to model the dosimetry of 225Ac its daughters [64]. This dosimetric evaluation of α-particle emitters required that all decays, including those of unstable intermediates be included in the calculation. These calculations were complicated by the differential biodistributions of each of the progeny due to their varied periodic properties. The formalism that was presented accounted for the known pharmacokinetic profiles of the daughters and the effective biodistribution focused on the site at which the 225Ac decayed. The cumulative decays of a daughter present in a particular tissue were estimated using a probability matrix which described the likelihood of daughter decay in a particular tissue as a function of the decay site of the parent.
Cellular dose conversion factors (DCF) were calculated employing the dose contributions of several progeny at the site of 225Ac decay that were made dependent on a threshold time parameter [65]. This enabled an estimate of the fraction of daughter decays expected at the site of parent decay. Previously tabulated S values (cell-surface to nucleus and cell-surface to cell) for each daughter were then scaled by this fraction and the sum over all progeny was performed to yield a cut-off time-dependent set of corresponding DCF values. These DCF values for the absorbed dose to the nuclear or cellular volume arising from cell-surface decays were presented as a function of the cut-off time for several different representative sets of cellular and nuclear dimensions. In contrast to the cellular S values that accounted only for the 225Ac decay, these cellular DCF values made it now possible include the contribution of progeny decays in cellular α-particle emitter dose calculations.
A theoretical estimate of the absorbed dose to key organs arising from the use of radionuclides with multiple unstable progeny was made using three sophisticated model features applied to different sized tumor masses and carrier specifications [66]. Each of the 225Ac progeny of has its own biodistribution profile and half-life, therefore, including their contributions would yield a more accurate prediction of absorbed dose and potential toxicity. The first model restricted the transport to a function that yielded either the place of origin or the place(s) of biodistribution depending on the half-life of the parent radionuclide. The second model included the transient time in the bloodstream and the third model incorporated additional binding at or within the tumor. (Note that the second model allowed for radionuclide decay and additional daughter production while transiting from one location to the next and that the third model relaxed the constraint that the residence time within the tumor was solely based on the half-life of the parent.) Calculations simulated were both a rapidly accessible small (0.1 g) tumor and a large (10 g) solid tumor. In addition, the effects of varying the carrier molecule purity and mass amount, as well as tumor cell antigen saturation were examined. The results indicated that there was a distinct advantage in using a parent radionuclide such as 225Ac, having a 10 d half-life and yielding 4 alpha particles per decay. Lower normal tissue doses resulted for a given tumor dose in comparison to those radionuclides yielding fewer alpha particles.
Clinical trials in humans
[225Ac]-lintuzumab (HuM195) is in use in an ongoing Phase I clinical trial at Memorial Sloan-Kettering Cancer Center. At press, the twelfth patient had been treated. Patient accrual continues. This Phase I clinical trial with [225Ac]-lintuzumab resulted from a period of clinical investigation of CD33-targeted therapy in patients with AML, mainly relapsed or refractory. Early studies were conducted with the murine form, M195, labeled with 131I in patients with minimal residual disease (50 – 70 mCi/m2) or to intensify therapy prior to bone marrow transplant (BMT) (120–230 mCi/m2). However, the detection of human anti-mouse antibodies in a fraction of patients precludes additional M195 treatments in the further course of the disease [67]. The humanized form of this antibody, HuM195, demonstrated a lack of immunogenicity and an increased affinity to CD33. Intensification of therapy prior to BMT might be achieved with the longer ranged beta-emitter yttrium-90 and further clinical studies where conducted with 90Y labeled HuM195 for myeloablation [65]. In contrast, in non-myeloablative regimens, CD33 negative stem cells have to be spared from the non-specific cross radiation. This led to the strategy that employed the alpha-emitting nuclide 213Bi labeled to HuM195. In a Phase I clinical trial, the anti-leukemic effect of [213Bi]-HuM195 was demonstrated in patients with relatively high tumor burden [18]. Because of the large tumor burden in AML, the Phase I/II study used a regimen wherein Cytarabine was given prior to the [213Bi]-HuM195 to effect some cytoreduction before alpha-particle therapy. This led to the first clinical trial using the longer-lived and more potent 225Ac in humans using the [225Ac]DOTA-HuM195 construct [69].